

Abstract
The 2019 novel coronavirus (2019‐nCoV) outbreak has caused a large number of deaths with thousands of confirmed cases worldwide, especially in East Asia. This study took an immunoinformatics approach to identify significant cytotoxic T lymphocyte (CTL) and B cell epitopes in the 2019‐nCoV surface glycoprotein. Also, interactions between identified CTL epitopes and their corresponding major histocompatibility complex (MHC) class I supertype representatives prevalent in China were studied by molecular dynamics simulations. We identified five CTL epitopes, three sequential B cell epitopes and five discontinuous B cell epitopes in the viral surface glycoprotein. Also, during simulations, the CTL epitopes were observed to be binding MHC class I peptide‐binding grooves via multiple contacts, with continuous hydrogen bonds and salt bridge anchors, indicating their potential in generating immune responses. Some of these identified epitopes can be potential candidates for the development of 2019‐nCoV vaccines.
Keywords: 2019‐nCoV, coronavirus, COVID‐19, epitope prediction, immunoinformatics, SARS‐CoV‐2
Highlights
Five CTL epitopes and eight B cell epitopes were computationally identified.
Epitope‐MHC‐I complexes were studied by molecular dynamics simulation.
Persistent H‐bonds and salt bridges were observed between epitopes and MHC‐I.
The identified epitopes can be a promising foundation for vaccine development.
1. INTRODUCTION
The 2019 novel coronavirus (2019‐nCoV; Family Coronaviridae), also known as the Wuhan coronavirus, has created a global emergency the likes of which had not been seen since the 2003 severe acute respiratory syndrome (SARS) outbreak. As of 27th January 2020, the death toll from the coronavirus stood at 80, with more than 2500 confirmed cases in China, as well as in 11 other countries.1
The virus, first identified in Wuhan, Hubei, China, causes a contagious, respiratory infection. The incubation period is reportedly 2 to 10 days, and studies are underway to determine whether the transmission is possible from asymptomatic individuals.1 Similar to other coronaviruses, 2019‐nCoV reportedly possesses a surface glycoprotein. In more established coronavirus pathogenesis, this protein has been shown to bind host cellular receptors and to mediate membrane fusion.2 More importantly, it has been described as a potential vaccine target in both SARS‐coronavirus (SARS‐CoV) and Middle East respiratory syndrome coronavirus (MERS‐CoV).3, 4
This study aimed to identify significant cytotoxic T cell (CTL) and B cell epitopes within the 2019‐nCoV surface glycoprotein using bioinformatic analysis. We also studied the potential interaction of the putative CTL epitopes with their corresponding representative major histocompatibility complex (MCH) class I supertypes most frequently observed in the Chinese population. We investigated hydrogen bonding, salt‐bridges, and residue‐residue contacts between CTL binding epitopes and the MHC class I molecules by molecular dynamics (MD) simulations.
2. METHODS
Amino acid sequence of the 1273 AA, viral surface glycoprotein was retrieved in FASTA format from NCBI Protein Database (YP_009724390.1). Because of the novel nature of 2019‐nCoV, we compared the glycoprotein's alignment with the spike glycoprotein of SARS‐Cov (UniProt: P59594) to ensure sequence similarity in EMBOSS Water pairwise sequence aligner.5
NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL) was used to identify CTL epitopes within the glycoprotein sequence.6 NetCTL uses NetMHC server's artificial neural networks (ANNs) to predict MHC binding, NetChop‐3.0 to predict C‐terminal cleavages and a weight matrix to calculate TAP transport efficiency to generate predictions. The scores of the three prediction methods are merged into a single score, which can be translated as sensitivity and specificity for the epitope‐MHC‐I binding event. We predicted MHC‐peptide‐binding for the following MHC class I supertypes—A2, A3, B7, B44, and B62. Together, these supertypes account for the majority of human leukocyte antigen A (HLA‐A) (76.5%) and HLA‐B (67.1%) distribution in the Chinese population.7 Epitopes with a combined score of >0.75 (sensitivity = 0.80, specificity = 0.97) were further confirmed in CTLPred (http://crdd.osdd.net/raghava/ctlpred),8 where a consensus scoring approach was taken with thresholds of 0.51 (ANN) and 0.36 (support vector machines).
The three highest‐scoring epitopes for each supertype were selected for analyses of binding affinity to their corresponding representative supertypes in NetMHCpan 4.0 server which predicted epitope‐MHC‐I binding using ANN trained on quantitative binding data and mass spectroscopy‐derived MHC eluted ligands (http://www.cbs.dtu.dk/services/NetMHCpan).9 Default threshold % ranks of 0.5 for strong binders, and 2 for weak binders were used.
Binding CTL epitopes were selected for homology modeling in PEP‐FOLD3, and structures with the lowest coarse‐grained energy were selected for protein‐protein docking with corresponding HLA chains using ZDOCK 3.0.2.10, 11 Complexes that scored the highest, based on interface atomic contact energy, shape complementarity, and electrostatics were selected for MD simulations in NAMD. Structures were solvated inside a TIP3P water box, and neutralized with Cl− and/or Na+, as required. Energy minimization was carried out in a series of conjugate gradients to ensure appropriate structure geometry. Equilibration with restrained solute atoms was performed in an NPT ensemble (310 K, 1 bar) with periodic boundary conditions, and constant temperature and pressure dynamics via Langevin dynamics and Nose‐Hoover Langevin piston respectively. Finally, free production runs, without positional restraints were carried out for 2 ns. Hydrogen bonds between epitopes and HLA chains were mapped in VMD with a two‐way donor‐acceptor distance of 3 Å and an angle of 20° as thresholds. Residue‐residue contacts between epitopes and HLA chains were visualized in a contact map matrix, and salt bridge‐forming residues were identified using the oxygen‐nitrogen cut‐off distance of 3.2 Å.
Sequential B cell epitopes within the surface glycoprotein were identified in BepiPred 2.0 (http://www.cbs.dtu.dk/services/BepiPred), and residues scoring above the default threshold of 0.5 were further confirmed in ABCPred, with default threshold of 0.51 and an overlapping filter (http://crdd.osdd.net/raghava/abcpred).12, 13 BepiPred 2.0 uses a random forest algorithm trained on epitope and non‐epitope amino acids validated from crystal structures, whereas ABCPred employs ANN using fixed‐length patterns trained on 700 B cell epitopes and 700 random non‐epitopes. To identify discontinuous B cell epitopes, we used Ellipro using the surface glycoprotein structure modeled in RaptorX, and the top five scoring epitopes were selected as significant.14, 15 The modeled structure was validated by the Ramachandran plot generated in RAMPAGE.16 Antigenicity of all identified epitopes was predicted using VaxiJen 2.0.17 Conservation of the sequential epitopes within spike proteins across bat coronavirus (Bt‐CoV, UniProt: A3EX94), SARS‐CoV, and MERS‐CoV (UniProt: K9N5Q8) were studied by multiple sequence alignment in Clustal Omega and visualized in NCBI Multiple Sequence Alignment Viewer.18
3. RESULTS
The surface glycoprotein of 2019‐nCoV was found to have 76.3% identity and 87.3% similarity with the spike glycoprotein of SARS‐CoV.
We identified five CTL epitopes in the surface glycoprotein, with all five epitopes classified as strong binders to their corresponding MHC class I supertype representative (Table 1). During MDs simulations, we observed the CTL epitopes to bind with the peptide‐binding groove of MHC class I molecules, which is typically used for antigen presentation. The relevant residues of the epitopes and HLA chains in contact were mapped in a contact map matrix (Figure 1A). We observed salt bridge‐forming residues positioned over the viral epitopes in three of the five complexes, as well as continuous hydrogen bonding between epitopes and HLA chains maintained throughout the simulation in all complexes (Figure 1B,C). Mutations in salt bridge‐forming amino acids have been shown to weaken TCR recognition of MHC class I‐antigen complexes.19 Also, the continuous formation of a set of hydrogen bonds between the MHC molecules and epitopes indicates an accommodating nature of the HLA chains to bind epitopes within the groove between the two helices.20 Our results are therefore optimistic of ideal antigen presentation to CTLs.
Table 1.
CTL epitopes identified in 2019‐nCoV surface glycoprotein and their corresponding MHC class I supertype representative
| Epitope | Epitope score—ANN/SVM | Antigenicity (score) | HLA (% rank) |
|---|---|---|---|
| YLQPRTFLL | 0.83/0.64 | Y (0.45) | HLA‐A*02:01 (0.01) |
| GVYFASTEK | 0.58/0.98 | Y (0.71) | HLA‐A*03:01 (0.00) |
| EPVLKGVKL | 0.73/0.61 | Y (1.23) | HLA‐B*07:02 (0.28) |
| VVNQNAQAL | 0.77/0.78 | Y (0.47) | HLA‐B*07:02 (0.78) |
| WTAGAAAYY | 0.82/0.54 | Y (0.63) | HLA‐B*15:01 (0.37) |
Abbreviations: 2019‐nCoV, 2019 novel coronavirus; ANN/SVM, artificial neural network/support vector machine; CTL, cytotoxic T lymphocyte; HLA, human leukocyte antigen; MHC, major histocompatibility complex.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 1.

Interactions between cytotoxic T lymphocyte binding epitopes and human leukocyte antigen (HLA) chains of corresponding major histocompatibility complex class I supertype representative during molecular dynamics simulations. A, Residue‐residue contacts (X: epitope, Y: HLA) were mapped in contact map matrices. B, Salt bridge‐forming residues were detected in three complexes. C, Continuous hydrogen bonds were observed between all epitopes and corresponding HLA chains throughout the simulations
We also identified three sequential B cell epitopes, and following modeling of 98% residues of the surface glycoprotein (P = 1.03e−15, uGDT = 946), we further identified five discontinuous B cell epitopes in the structure (Tables 2 and 3). Ramachandran plot indicated 94.2% (1194 AA) residues in the favored region, 5% (63 AA) residues in the allowed region, and 0.8% (10 AA) residues in the outlier region (Figure 2).
Table 2.
Sequential B cell epitopes identified in 2019‐nCoV surface glycoprotein
| Epitope | Epitope probability | Antigenicity (score) | IFN‐γ epitope (score) |
|---|---|---|---|
| CVNLTTRTQLPPAYTN | 0.74 | Y (1.38) | N (−0.92) |
| NVTWFHAIHVSGTNGT | 0.55 | Y (0.84) | N (−0.30) |
| SFSTFKCYGVSPTKLNDL | 0.69 | Y (1.06) | N (−0.16) |
Abbreviations: 2019‐nCoV, 2019 novel coronavirus; IFN‐γ, interferon γ.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 3.
Discontinuous B cell epitopes identified in 2019‐nCoV surface glycoprotein
| Sl. no | Epitope:position | Score |
|---|---|---|
| 1 | H1271, Y1272, T1273 | 0.998 |
| 2 | E1258, D1259, D1260, S1261, E1262, P1263, V1264, L1265, K1266, G1267, V1268, K1269, L1270 | 0.985 |
| 3 | G838, D839, C840, L841, G842, D843, I844, A845, A846, R847, D848, L849, I850, C851, A852, Q853, K854, F855 | 0.866 |
| 4 | Y1215, I1216, L1218, G1219, F1220, I1221, A1222, G1223, L1224, I1225, A1226, I1227, V1228, M1229, V1230, T1231, I1232, M1233, L1234, C1235, C1236, M1237, T1238, S1239, C1240, C1241, S1242, C1243, L1244, K1245, G1246, C1247, C1248, S1249, C1250, G1251, S1252, C1253, C1254, K1255, F1256, D1257 | 0.832 |
| 5 | M1, F2, V3, F4, L5, V6, L7, L8, P9, L10, V11, S12, S13, Q14, C15, V16, N17, L18 | 0.786 |
Abbreviations: 2019‐nCoV, 2019 novel coronavirus; Bt‐CoV, bat coronavirus; CTL, cytotoxic T lymphocyte; MERS‐CoV, Middle East respiratory syndrome coronavirus; SARS‐CoV, severe acute respiratory syndrome coronavirus.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 2.
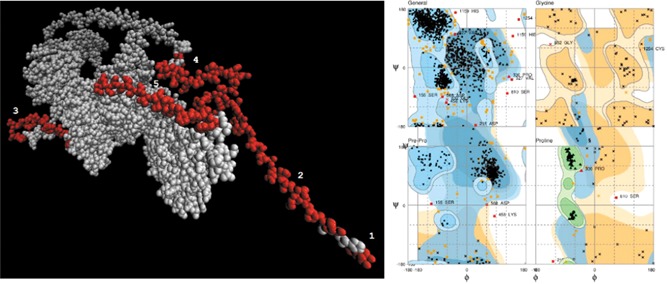
Discontinuous B cell epitopes in 2019‐nCoV surface glycoprotein and structure validation by Ramachandran plot. Left—five discontinuous B cell epitopes identified in the modeled structure, detailed in Table 3. Right—ramachandran plot indicated a high proportion of residues to be located within the favored and allowed regions. 2019‐nCoV, 2019 novel coronavirus
With the exception of CTL epitope—VVNQNAQAL, which was found to have 100% identity with SARS‐CoV, all identified sequential CTL and B cell epitopes were at least partially unique to 2019‐nCoV, compared with Bt‐CoV, SARS‐CoV, and MERS‐CoV. CTL epitope—EPVLKGVKL did not match any corresponding sequences of the other coronaviruses (Figure 3).
Figure 3.

Visualization of frequency‐based differences in residues of sequential CTL and B cell epitopes across bat coronavirus, MERS‐coronavirus and SARS‐coronavirus. CTL, cytotoxic T lymphocyte; MERS, Middle East respiratory syndrome; SARS, severe acute respiratory syndrome
4. DISCUSSION
Rapid identification of immune epitopes is of crucial importance at the time of an impending pandemic. In this study, we attempted to identify significant CTL and B cell epitopes in the surface glycoprotein of 2019‐nCoV.
CTLs can become activated after they detect infected cells presenting the offending viral antigens as part of surface antigen‐MHC‐I complexes. Successful presentation depends on the effective harboring of antigens by MHC‐I molecules via hydrogen bonds and salt bridge interactions that allow both high affinity with broader specificity.21 After docking and structure refinement, we analyzed interactions between individual CTL epitopes and HLA chains of their corresponding MHC class I supertype representative by MDs simulations. We observed multiple, maintained hydrogen bond interactions, in addition to the presence of salt‐bridges in three complexes. Collectively, these interactions indicate a potentially successful presentation of the epitopes by MHC class I molecules, thereby activating CTLs and generating immune responses. We compared the epitopes obtained in our study with epitopes predicted in the spike protein of MERS‐CoV and SARS‐CoV.22, 23 We found one overlapping CTL epitope between MERS‐CoV and 2019‐nCoV with one gap and one mismatch (YLQPRTFLL/YKLQPLTFLL), and no comparable epitopes with SARS‐CoV. In addition to the activation of CTLs, successful immunogens need to generate a persistent humoral immunity and in our study, we identified three sequential and five discontinuous B cell epitopes in the surface glycoprotein.
There are a few limitations of our study. Epitope detection in glycoproteins is often complicated by the presence of covalently attached glycans. Modeling viral glycoproteins with the associated glycans is especially challenging due to the lack of related structures, although attempts have been made in hepatitis C and rabies.24, 25 Second, similar to other RNA virus, coronavirus replication is error‐prone, with a reported mutation rate of 4 × 10−4 substitutions per site per year in SARS‐CoV.26 This necessitates urgency in antiviral vaccine development before the identified epitopes are potentially rendered obsolete.
Baruah V, Bose S. Immunoinformatics‐aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019‐nCoV. J Med Virol. 2020;92:495–500. 10.1002/jmv.25698
REFERENCES
- 1. World Health Organization . Novel coronavirus (2019‐nCoV) situation report‐7. Report 2020. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200127-sitrep-7-2019--ncov.pdf
- 2. Li F. Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology. 2016;3:237‐261. 10.1146/annurev-virology-110615-042301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Du L, He Y, Zhou Y, Liu S, Zheng B‐J, Jiang S. The spike protein of SARS‐CoV—a target for vaccine and therapeutic development. Nat Rev Microbiol. 2009;7:226‐236. 10.1038/nrmicro2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou Y, Jiang S, Du L. Prospects for a MERS‐CoV spike vaccine. Expert Rev Vaccines. 2018;17:677‐686. 10.1080/14760584.2018.1506702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith T, Waterman M. Identification of common molecular subsequences. J Mol Biol. 1981;147:195‐197. 10.1016/0022-2836(81)90087-5 [DOI] [PubMed] [Google Scholar]
- 6. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large‐scale validation of methods for cytotoxic T‐lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424 10.1186/1471-2105-8-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. dos Santos Francisco R, Buhler S, Nunes JM, et al. HLA supertype variation across populations: new insights into the role of natural selection in the evolution of HLA‐A and HLA‐B polymorphisms. Immunogenetics. 2015;67:651‐663. 10.1007/s00251-015-0875-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhasin M, Raghava G. Prediction of CTL epitopes using QM, SVM, and ANN techniques. Vaccine. 2004;22:3195‐3204. 10.1016/j.vaccine.2004.02.005 [DOI] [PubMed] [Google Scholar]
- 9. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan‐4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. The Journal of Immunology. 2017;199:3360‐3368. 10.4049/jimmunol.1700893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP‐FOLD approach for peptide and miniprotein structure prediction. J Chem Theory Comput. 2014;10:4745‐4758. 10.1021/ct500592m [DOI] [PubMed] [Google Scholar]
- 11. Pierce BG, Wiehe K, Hwang H, Kim B‐H, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein‐protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771‐1773. 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred‐2.0: improving sequence‐based B‐cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45:45‐W29. 10.1093/nar/gkx346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saha S, Raghava GPS. Prediction of continuous B‐cell epitopes in an antigen using recurrent neural network. Proteins: Struct, Funct, Bioinf. 2006;65:40‐48. 10.1002/prot.21078 [DOI] [PubMed] [Google Scholar]
- 14. Ponomarenko J, Bui H‐H, Li W, et al. ElliPro: a new structure‐based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514 10.1186/1471-2105-9-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Källberg M, Wang H, Wang S, et al. Template‐based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511‐1522. 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lovell SC, Davis IW, Arendall WB, et al. Structure validation by Cα geometry: ϕ, ψ, and Cβ deviation. Proteins: Struct, Funct, Bioinf. 2003;50:437‐450. 10.1002/prot.10286 [DOI] [PubMed] [Google Scholar]
- 17. Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens, and subunit vaccines. BMC Bioinformatics. 2007;8:8 10.1186/1471-2105-8-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji W, Niu L, Peng W, et al. Salt bridge‐forming residues positioned over viral peptides presented by MHC class I impacts T‐cell recognition in a binding‐dependent manner. Mol Immunol. 2019;112:274‐282. 10.1016/j.molimm.2019.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wieczorek M, Abualrous ET, Sticht J, et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8:8 10.3389/fimmu.2017.00292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rognan D, Zimmermann N, Jung G, Folkers G. Molecular dynamics study of a complex between the human histocompatibility antigen HLA‐A2 and the IMP58‐66 nonapeptide from influenza virus matrix protein. Eur J Biochem. 1992;208:101‐113. 10.1111/j.1432-1033.1992.tb17163.x [DOI] [PubMed] [Google Scholar]
- 22. Qamar MTU, Saleem S, Ashfaq UA, Bari A, Anwar F, Alqahtani S. Epitope‐based peptide vaccine design and target site depiction against Middle East respiratory syndrome coronavirus: an immune‐informatics study. J Transl Med. 2019;17:362 10.1186/s12967-019-2116-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang J, Cao Y, Bu X, Wu C. Residue analysis of a CTL epitope of SARS‐CoV spike protein by IFN‐gamma production and bioinformatics prediction. BMC Immunol. 2012;13:50 10.1186/1471-2172-13-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guest JD, Pierce BG. Computational modeling of hepatitis C virus envelope glycoprotein structure and recognition. Front Immunol. 2018;9, 10.3389/fimmu.2018.01117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fernando B‐G, Yersin C‐T, José C‐B, Paola Z‐S. Predicted 3D model of the rabies virus glycoprotein trimer. BioMed Res Int. 2016;2016:1‐11. 10.1155/2016/1674580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salemi M, Fitch WM, Ciccozzi M, Ruiz‐Alvarez MJ, Rezza G, Lewis MJ. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J Virol. 2004;78:1602‐1603. 10.1128/jvi.78.3.1602-1603.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]