

Abstract
The proprotein convertases (PCs) furin, PC5, PACE4, and PC7 cleave secretory proteins after basic residues, including the HIV envelope glycoprotein (gp160) and Vpr. We evaluated the abundance of PC mRNAs in postmortem brains of individuals exhibiting HIV-associated neurocognitive disorder (HAND), likely driven by neuroinflammation and neurotoxic HIV proteins (e.g., envelope and Vpr). Concomitant with increased inflammation-related gene expression (interleukin-1β [IL-1β]), the mRNA levels of the above PCs are significantly increased, together with those of the proteinase-activated receptor 1 (PAR1), an inflammation-associated receptor that is cleaved by thrombin at ProArg41↓ (where the down arrow indicates the cleavage location), and potentially by PCs at Arg41XXXXArg46↓. The latter motif in PAR1, but not its R46A mutant, drives its interactions with PCs. Indeed, PAR1 upregulation leads to the inhibition of membrane-bound furin, PC5B, and PC7 and inhibits gp160 processing and HIV infectivity. Additionally, a proximity ligation assay revealed that furin and PC7 interact with PAR1. Reciprocally, increased furin expression reduces the plasma membrane abundance of PAR1 by trapping it in the trans-Golgi network. Furthermore, soluble PC5A/PACE4 can target/disarm cell surface PAR1 through cleavage at Arg46↓. PACE4/PC5A decreased calcium mobilization induced by thrombin stimulation. Our data reveal a new PC-PAR1-interaction pathway, which offsets the effects of HIV-induced neuroinflammation, viral infection, and potentially the development of HAND.
INTRODUCTION
The mammalian proprotein convertases (PCs) form a family of nine secretory serine proteinases exhibiting similarities to bacterial subtilisin and to kexin of Saccharomyces cerevisiae (1, 2).There are seven basic amino acid-specific PCs more specifically related to yeast kexin: PC1 (also known as PC1/3), PC2, furin, PC4, PC5 (also known as PC5/6), PACE4, and PC7. These proteinases cleave after single or paired basic amino acids within the motif (R/K)-Xn-(R)↓, where Xn is 0, 2, 4, or 6 variable amino acids separating the two canonical basic residues required for cleavage recognition and the down arrow indicates the cleavage location.
The membrane-bound furin, PC5B, and PC7 and soluble PC5A and PACE4 have a wide tissue distribution and proteolytically process precursors in the constitutive secretory pathway. These PCs are implicated in the activation of a wide variety of proteins such as growth factors, receptors, enzymes, blood coagulation factors, and viral envelope glycoproteins (2). Furin has enzymatic activity primarily in the trans-Golgi network (TGN) and at the cell surface and/or endosomes (3), whereas in many instances the soluble isoform PC5A and PACE4 exert their functions either at the cell surface as they bind heparin sulfate proteoglycans (HSPGs) or extracellularly (1, 4). PC7 has been reported to function exclusively near the cell surface (5, 6). Although a number of endogenous substrates of PCs are known, the potential role of PCs in targeting pathogen proteins has also been reported (2, 7). For example, the envelope glycoprotein gp160 (8, 9) and the cytotoxic accessory protein Vpr (10) involved in the pathophysiology of the human immunodeficiency virus (HIV) are potential targets of the convertase family of proteinases.
HIV infects the central nervous system soon after primary infection (11) and establishes a tissue reservoir by productively infecting brain myeloid cells, which eventually leads to neurocognitive impairment in a subset of patients (reviewed in reference 12). HIV-associated neurocognitive disorder (HAND) represents a daunting and ongoing challenge in the diagnosis and care of people with HIV/AIDS. HAND displays a spectrum of cognitive and motor symptoms, including loss of memory, reduced concentration, impaired decision making, and psychomotor slowing (13, 14). The clinical manifestations of HAND result from concurrent neurodegeneration associated with synapto-dendritic injury and neuronal death. This process is believed to be driven by neuroinflammation involving innate immune activation of infiltrating macrophages and resident microglia within the brain, coupled with the direct neurotoxic effects of secreted HIV-encoded proteins (15), including Vpr (16) and gp120 (17).
Highly active antiretroviral therapy (HAART), which has been demonstrated to be effective in controlling viral replication, decreased the incidence of HAND although its prevalence is increasing due to improved survival. Suppression of viral replication by HAART delays, and perhaps prevents, the onset of HAND in patients with HIV infection. Nonetheless, a substantial proportion of HIV/AIDS patients receiving HAART display HAND (∼20%), with a worsened overall survival (18). HIV/AIDS patients receiving HAART at the time of death display both provirus and viral RNA together with ongoing neuroinflammation (19).
Neuroinflammation and neurodegeneration have been linked to the upregulation of proteinase-activated receptor 1 (PAR1) and its downstream inflammatory signaling (20, 21). PAR1 is a G-protein coupled receptor that is highly expressed in astrocytes, microglia, and neurons. Its expression is also significantly upregulated at the mRNA and protein levels in astrocytes of brain samples of HIV/AIDS patients (22). PAR1 downstream signaling is activated via cleavage by thrombin, a blood coagulation factor (23), in its N-terminal extracellular domain at Arg41↓. This cleavage exposes a new N-terminal sequence composed of amino acids (aa) 42 to 47, SFLLRN47, that acts as an intramolecular ligand, which binds the extracellular loop located between transmembrane domains 2 and 3 of PAR1, and induces downstream signaling leading to inflammation (21). In addition, cleavage of the PAR1 N-terminal sequence at Arg46↓, e.g., by activated protein C (APC), unmasks an alternate PAR1-regulated pathway that triggers anti-inflammatory protective signaling by PAR1 (24–26). This Arg46↓ cleavage by APC also disarms PAR1 by separating the thrombin target sequence (ProArg41↓) from the receptor so that thrombin can no longer activate inflammatory signaling.
Of importance for our present study, we noticed that the PAR1-tethered ligand sequence (underlined), LDPR41SFLLR46NPN, exhibits a potential PC cleavage motif (position 1 [P1] Arg46 and P6 Arg41 in PAR1; in boldface) (2). Interestingly, a recent report demonstrated that PAR1 inhibits furin-mediated processing of the human metapneumovirus glycoprotein Fc (27). However, the underlying mechanism, the implication of other PCs, and their ability to regulate PAR1 signaling and its cellular trafficking are unknown. Because PCs can potentially target not only PAR1 but also the gp160 envelope glycoprotein of HIV (28–30), we hypothesized that, along with PAR1 (22), the levels of PCs could be altered in the setting of HAND. Therefore, our work sought to (i) evaluate mRNA expression levels of furin, PC5, PACE4, and PC7 in postmortem brain samples from individuals with HIV/AIDS and (ii) assess the processing of PAR1 by one or more of the above PCs to determine whether this cleavage could regulate its function.
MATERIALS AND METHODS
Quantitative reverse transcription-PCR (RT-PCR) analysis.
Using TRIzol (Invitrogen) and an RNeasy purification kit (Qiagen), total RNA was prepared from frontal cortical samples from HIV-uninfected patients (group A; n = 10), HIV-infected patients without neurocognitive impairment (group B; n = 10), HIV-infected patients with neurocognitive impairment (group C; n = 10), and HIV-infected patients with neurocognitive impairment and encephalitis (group D; n = 10). HeLa cells cultured in serum-free medium were treated with a cytokine cocktail composed of tumor necrosis factor alpha (TNF-α; 50 ng/ml), gamma interferon (IFN-γ; 50 ng/ml), and interleukin-1β (IL-1β; 10 ng/ml) for 24 h before they were collected for RNA extraction in TRIzol. The cDNAs were synthesized using Superscript II reverse transcriptase (Invitrogen). Semiquantitative real-time PCR was performed using SYBR green (IQ SYBR Supermix; Bio-Rad) detection and the ΔCT (where CT is threshold cycle) method (31). Threshold cycle values for the gene of interest were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (for brain samples) and the TATA binding protein (for cell lysates) and are represented as the mean relative fold change compared to control sample values. Excel software was used for calculating standard errors of the means (SEM) and Student's t test probabilities.
Immunohistochemistry.
Human brain sections (8 μm) were deparaffinized and hydrated using decreasing concentrations of ethanol. Sections were boiled in 0.01 M citrate buffer, pH 6.0, for 10 min for PAR1 and glial fibrillary acidic protein (GFAP) immunostaining. Endogenous peroxidases were blocked by incubating sections in 0.3% hydrogen peroxide for 20 min. To prevent nonspecific binding, sections were preincubated with 10% normal goat serum–0.5% Triton X-100 for 1 h at room temperature. Rabbit anti-human PAR1 antiserum was used in accordance with previously described work (22). Mouse anti-GFAP antibodies were purchased from Dako (Copenhagen, Denmark). Primary antibodies were diluted in phosphate-buffered saline (PBS)–serum (PAR1, 1:500; GFAP, 1:1,000) and incubated overnight at room temperature, followed by washing. All washes were conducted for 15 min with 0.01 M PBS, pH 7.4, and antibodies were diluted in PBS containing 10% normal goat serum. Immunolabeling with primary antibodies was detected with biotinylated goat anti-rabbit or biotinylated goat anti-mouse antibodies (Vector Laboratories) and then with avidin-biotin-peroxidase complexes (Vector Laboratories) for 1 h at room temperature for each step. Immunoreactivity was detected using 3,3′-diaminobenzidine tetrachloride (brown) and/or 5-bromo-4-chloroindolylphosphate (blue) (32). All human brain specimens were collected with consent (19).
In situ hybridization.
Ten-micrometer-thick cryosections were prepared from brains of 3-month-old mice, fixed in 4% formaldehyde, and hybridized as previously described (33) with mouse sense (negative control) and antisense cRNA probes. The latter probes corresponded to the mouse PAR1, furin, PC5, PACE4, or PC7 coding region for aa 1 to 420, 1 to 793, 80 to 348, 1 to 214, 1 to 213, respectively, and were synthesized using 35S-UTP (PerkinElmer).
In vitro assays.
Enzymatic in vitro activities of the purified soluble furin, PC5/6, PACE4, and PC7 (a gift of Robert Day, Université de Sherbrooke) were measured at 37°C in 100 μl of buffer (25 mM Tris-HCl, 1 mM CaCl2, pH 7) in the presence of a 100 μM concentration of the fluorogenic substrate pyroglutamic acid-RTKR-7-amido-4-methylcoumarin (Pyr-RTKR-MCA; Peptide International). The release of free 7-amino-4-methyl-coumarin (AMC) was detected with a Spectra Max Gemini EM microplate spectrofluorimeter (Molecular Devices) (excitation, 370 nm; emission, 460 nm; emission cutoff, 435 nm). One unit of enzymatic activity was defined as 1 pmol of AMC released from 100 μM Pyr-RTKR-MCA/min at 37°C. The 19-mer synthetic peptide mimicking the tethered ligand and putative PC cleavage site (in boldface) of human PAR1, NATLDPR41SFLLR46↓NPNDKYE (hPAR135–53), was obtained from GenScript. To assess the in vitro cleavage of hPAR135–53 by PCs, the synthetic peptide was incubated at 37°C in vitro (100 μM peptide in 100 μl of buffer consisting of 25 mM Tris-morpholineethanesulfonic acid [MES], 1 mM CaCl2, pH 7) with 90 nM active purified PCs (18 h) or with 2 μg/ml trypsin (5 min and 1 h). Trypsin cleaves at Arg41↓ (preferentially) and at Arg46↓ and was used as a reference for the reverse-phase high-pressure liquid chromatography (RP-HPLC) profile of the resulting peptide fragments; a 5-min digestion results in cleavage at Arg41↓ and a 1-h digestion leads to cleavage at both Arg41↓ and Arg46↓. The digestion products were separated by RP-HPLC on a Varian C18 column (particle size, 5 μm; pore size, 100 Å; 4.6 by 250 mm) using a 5% to 50% acetonitrile (plus 0.1% trifluoroacetic acid [TFA]) gradient over 45 min at a flow rate of 1 ml/min. Peptide bonds were detected at 214 nm, and Tyr-containing peptides were detected at 280 nm. By comparing the RP-HPLC profiles of trypsin- and PC-digested hPAR135–53, it was determined that PACE4 cleaves the 19-mer at the predicted site R41SFLLR46↓. The percent cleavage was calculated as the ratio of the normalized peak area (peak area/number of peptide bonds) of the C-terminal fragment NPNDKYE to that of the intact 19-mer peptide (at time zero).
Plasmids and reagents.
The cDNA encoding human PAR1 (hPAR1; pcDNA3.1 vector containing mCherry at the N terminus and enhanced yellow fluorescent protein [eYFP] at the C terminus) was used as reported previously (34). Site-directed mutagenesis was used to generate the human PAR1 mutants R46A and R41A and the double mutant R41A R46A. All PC constructions (human furin, mouse PC5A, mouse PC5B, human PACE4, and human PC7) were cloned in pcDNA3 or pIRES2-EGFP (where IRES is internal ribosome entry site and EGFP is enhanced green fluorescent protein) vectors, as previously described (35). Plasmid expressors encoding hemagglutinin (HA)-tagged Vpr as well as HxBc2 Env were previously described (10, 36). The plasmids coding for mouse growth differentiating factor 11 (GDF11) (37) and human transferrin receptor 1 (TfR1) (5) were used as reported previously.
Cell culture and cDNA transfections.
HEK293 (human embryonic kidney-derived epithelial) and COS-1 (monkey kidney fibroblast) cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) (Invitrogen) and were maintained at 37°C under 5% CO2. Using JetPrime (PolyPlus) for HEK293 and Lipofectamine 2000 (Invitrogen) for COS-1 (unless otherwise indicated), 80 to 90% confluent cells were transiently transfected with pcDNA3 recombinants of dually tagged human PAR1 with full-length human furin, mouse PC5A or PC5B, human PACE4, human PC7, or an empty pcDNA3 plasmid. Twenty-four hours after transfection, the cells were washed and incubated in serum-free medium for an additional 20 h before medium collection and cell lysis.
Immunofluorescence.
HEK293 cells were plated on polylysine-coated coverslips and transfected. Twenty-four hours posttransfection, fresh serum-free medium with 1 unit/ml of thrombin (Calbiochem) and/or 5 μM E64 (a cysteine proteinase inhibitor; Calbiochem) was added to the cells and incubated overnight. Our previous work (34) indicated that many cells, including HEK293 cells, secrete PAR-regulating proteinases that can cleave the N terminus of PARs, including PAR1. Thus, E64 was found to preserve the N terminus so that it can in principle be targeted by other noncysteine proteinase enzymes. In experiments using PC-specific inhibitors, 25 μM cell-permeable decanoyl-RVKR-chloromethylketone (cmk) (RVKR-cmk; Bachem Bioscience) and 10 μM nonpermeable hexa-d-arginine (D6R; EMD Chemicals) were added at this point with fresh serum-free medium (38). After 20 h, the cells were washed three times with PBS and then fixed in 3.7% formaldehyde with 0.01 mM sucrose for 15 min at room temperature. Following a final three washes with PBS, the coverslips were mounted with ProLong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen). Immunofluorescence analyses were performed with a Zeiss LSM-710 confocal microscope. When antibodies were used, the cells were washed three times with PBS and permeabilized with 0.25% Triton X-100 (or with PBS for nonpermeabilized cells) for 7 min following fixation. Blocking was performed subsequent to an additional three washes with PBS using 1% bovine serum albumin for 30 min. The cells were incubated overnight at 4°C with anti-golgin-97 monoclonal antibody (MAb) (Santa Cruz), V5 MAb (Invitrogen) for human furin, and monoclonal HA (in-house) primary antibodies for human PACS1 at 1:250 dilutions in 1% bovine serum albumin. Alexa Fluor 647-conjugated secondary antibodies (Invitrogen), diluted 1:500 in 1% bovine serum albumin, were incubated for 1 h at room temperature after three washes with PBS.
Western blot analyses.
Proteins were extracted from cells in 50 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% SDS, 1% Nonidet P40, and 0.25% Na deoxycholate (1× radioimmunoprecipitation assay [RIPA] buffer) in the presence of a cocktail of protease inhibitors (Roche). Protein concentrations were estimated by a Bradford assay. After SDS-PAGE and blotting onto polyvinylidene difluoride (PVDF) membranes, the latter were incubated with Chessie 8 anti-gp41 (1:10) (39), monoclonal HA (1:1,000; American Type Culture Collection), monoclonal V5 (1:5,000; Invitrogen), monoclonal Flag M2 (1:5,000; Stratagene), and rabbit polyclonal anti-β-actin (1:1,000; Sigma-Aldrich) antibodies and finally with appropriate secondary antibodies. Immunoreactive species were detected using enzymatic chemiluminescence (Amersham Bioscience) and quantified using ImageJ, version 1.41, software (NIH).
Calcium mobilization assay.
HEK293 cells from a T75 flask were detached in enzyme-free, EDTA-containing, cell dissociation buffer and resuspended in serum-containing growth medium, and approximately 100,000 cells/well were seeded into cell culture-treated clear-bottom 24-well CellBIND plates (Corning) coated with polylysine. The next day, cells were transfected with 0.5 μg of the PACE4 and PC5A cDNAs per well with a FuGene transfection reagent according to the manufacturer's protocol. At 24 h posttransfection the medium was replaced with serum-containing growth medium. The next day the culture medium was replaced with isotonic Hanks buffered salt solution (HBSS) and fluo-4-acetoxymethyl ester (AM) no-wash calcium indicator dye (Invitrogen), followed by incubation at 37°C for 30 min. Intracellular fluorescence (excitation at 494 nm and emission at 516 nm) was monitored for 2 min after the addition of 20 nM thrombin, using n Envision 2104 Multilabel Reader (PerkinElmer Life Sciences). The relative fluorescence measured per second was plotted, and the average values from each experiment for each condition were analyzed utilizing a nonlinear regression equation (GraphPad Prism). Peak height was calculated from the highest peak point to baseline of the average nonlinear regression lines.
PLA.
HEK293 cells seeded into 24-well plates were transfected with cDNAs with a FuGene transfection reagent according to the manufacturer's protocol and 24 h later reseeded into a μ-Slide eight-well ibiTreat microscopy chamber (Research Products/Ibidi). Cells were incubated with rabbit anti-PAR1 (Santa Cruz) and mouse anti-V5 (Invitrogen) primary antibodies overnight at 4°C. Duolink in situ (Olink Bioscience) proximity ligation assay (PLA) probes, anti-rabbit antibody minus sense and anti-mouse antibody plus sense, with a far-red detection kit were utilized to detect the interaction of mCherry-hPAR1-eYFP with V5-tagged furin or PC7 according to the Duolink in situ protocol. The number of dots (indicating interactions of PLA probes) per cell were manually counted utilizing ImageJ software (NIH).
PAR1 inhibition of gp160 processing and viral infectivity.
For protein expression assays, 1 × 106 HEK293T cells were seeded in 100-mm plates and transfected using the calcium phosphate procedure with 5 μg of HIV-1 NL4-3 proviral DNA (NIH AIDS Reagent Program, NIH) (40) and either 1 μg of control vector EGFP-C1 (Clontech) or combinations of control vector and wild-type (WT) mCherry-hPAR1-eYFP or mutant mCherry-hPAR1 R46A-eYFP constructs. Cells were harvested after 2 days of culture and lysed in buffer (25 mM Tris, pH 7.4, 1% Triton X-100, 150 mM NaCl) containing protease inhibitors (Roche). Cell supernatants were briefly centrifuged and filtered on 0.45-μm-pore-size membranes, and virus pellets were obtained following ultracentrifugation on 20% (wt/vol) sucrose cushions for 2 h at 130,000 × g in a Beckman ultracentrifuge. Virus pellets were then resuspended in 100 μl of PBS and aliquoted. A total of 20 μg of protein in cell lysates was added to 5× Laemmli buffer, and the proteins were separated by 12.5% SDS-PAGE prior to transfer on nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% milk in Tris-buffered saline (TBS)–0.1% Tween 20, washed in TBS, and incubated with the appropriate antibodies overnight at 4°C: mouse anti-gp160 (Chessie 13-39.1, 1:10) (39), mouse anti-gp41 (Chessie 8, 1:10) (39), and mouse anti-gp120 (IIIB 902, 1:10) (41, 42) (all obtained from the NIH AIDS Reagent Program); mouse anti-p24 (31-90-25, 1:3,000; ME Biotech Services); rabbit anti-GFP (1:500; Life Technologies); and rabbit anti-GAPDH (1:1,000; Biolegend). Following three washes in TBS–0.1% Tween 20, blots were incubated with anti-mouse or anti-rabbit horseradish peroxidase (HRP)-labeled antibodies (1:5,000), washed, and processed for enhanced chemiluminescence assay (Pierce). Films were scanned, and protein signals were quantified using ImageJ (NIH) software. For infection assays, the TZM-bl reporter cell line (obtained from the NIH AIDS Reagent Program) (43, 44) was used. These HeLa-based modified cells harbor a copy of the firefly luciferase gene under the control of the HIV-2 long terminal repeat (LTR) and express CD4 and the HIV coreceptors CXCR4 and CCR5 (43). Following HIV-1 p24 quantitation by enzyme-linked immunosorbent assay (ELISA; XpressBio Life Science), virus obtained from the previously described HEK293T cells cotransfected with 0.1 μg of WT or mutant R46A PAR1 was used in the assay. Briefly, either 1 ng, 0.2 ng, or 0.04 ng of virus was added to 30,000 TZM-bl cells/well in 24-well plates. Following 2 days of culture, cells were lysed, and luciferase activity was measured using a Promega luciferase assay kit (Promega) on a GloMax luminometer. Infections using the most diluted virus (0.04 ng) were nonsaturating and were used to determine viral infectivity.
Ethics approvals.
The decedents used in this work were from the National NeuroAIDS Tissue Consortium (NNTC) (19). The use of autopsied brain tissues is part of ongoing research (Pro00002291) approved by the University of Alberta Human Research Ethics Board (Biomedical). Written informed consent documents were signed for all samples collected. The protocols for obtaining postmortem brain samples comply with all federal and institutional guidelines, with special respect for the confidentiality of the donor's identity, and were approved by the ethics committee of the University of Texas Medical Branch, Galveston.
Statistical analysis.
Quantifications are defined as means ± SEM and expressed as percentages of the control value, which was considered to be 100%. The statistical significance was evaluated by Student's t test. Probability values of <0.05 were considered significant. Data were analyzed and graphics were generated with the Prism program (GraphPad Software).
RESULTS
Furin, PC5, PACE4, and PC7 expression levels are upregulated along with PAR1 in the brains of HAND patients with neuroinflammation.
The frontal cortex is a particularly vulnerable region of the brain in HAND (11). Thus, to test our hypothesis that PCs are elevated in brains of HIV-infected subjects, we measured PC-expression in the frontal cortex from HIV-negative and HIV-positive (HIV− and HIV+, respectively) individuals with neurocognitive impairment and encephalitis (n = 10 for each group). Brain transcript analyses revealed that furin, PC5, PACE4, and PC7 mRNA levels were all markedly increased (3- to 8-fold) in HIV+ brains of patients with neurocognitive impairment and encephalitis (HAND with encephalitis, or HIVE; group D), suggesting a role for PCs in neuroinflammation caused by HIV infection (Fig. 1A). Similar increases were observed in the expression levels of a prototypic inflammatory factor, IL-1β (10-fold), as well as those of PAR1 (6-fold), an inflammation-associated receptor that has previously been reported to be upregulated in HIVE (22). This G protein-coupled receptor (GPCR) has previously been linked to neuroinflammation and neurodegeneration (20–22). In contrast, the mRNA levels of PCs, IL-1β, and PAR1 did not change in HIV− brains (group A), HIV+ brains without neurocognitive impairment (group B), and HIV+ brains with neurocognitive impairment (HAND; group C) (Fig. 1A). Notably, the mRNA levels of inflammation-modulated caspase-4 and IL-33 from the same cohort were not affected (Fig. 1A), affirming that the increases observed were specific to HIV-induced neuroinflammation and not to a general inflammatory response.
FIG 1.

Expression of PCs in human HIV-positive brain and cells undergoing inflammation. (A) mRNA levels of PCs were measured in brains from patients with HIV/AIDS (HIV+) (group B, without neurocognitive impairment; group C, with neurocognitive impairment; group D, with neurocognitive impairment and encephalitis) using quantitative PCR analysis and compared to brains from of HIV-uninfected patients (HIV−) (group A, other disease controls; n = 10 per group). (B) PC expression was also measured in HeLa cells after 24 h of exposure to several cytokines (TNF-α, IL-1β, and IFN-γ). TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IFN-γ, gamma interferon (n = 3 per condition). *, P < 0.05; **, P < 0.005. Bars represent the means ± SEM, and P was determined by a Student t test (two-tailed). Casp4, caspase 4.
Additionally, an increased density of hypertrophied astrocytes (Fig. 2B) and microglia (Fig. 2D), which are widely assumed to be indicative of neuroinflammation, was evident in HIVE brain in contrast to observations in HIV− patient brain sections (Fig. 2A and C). PAR1 expression was markedly upregulated (Fig. 2F) in HIVE compared to levels in HIV− brain sections (Fig. 2E). This finding was limited to astrocytes and not apparent in neurons or microglia (data not shown).
FIG 2.
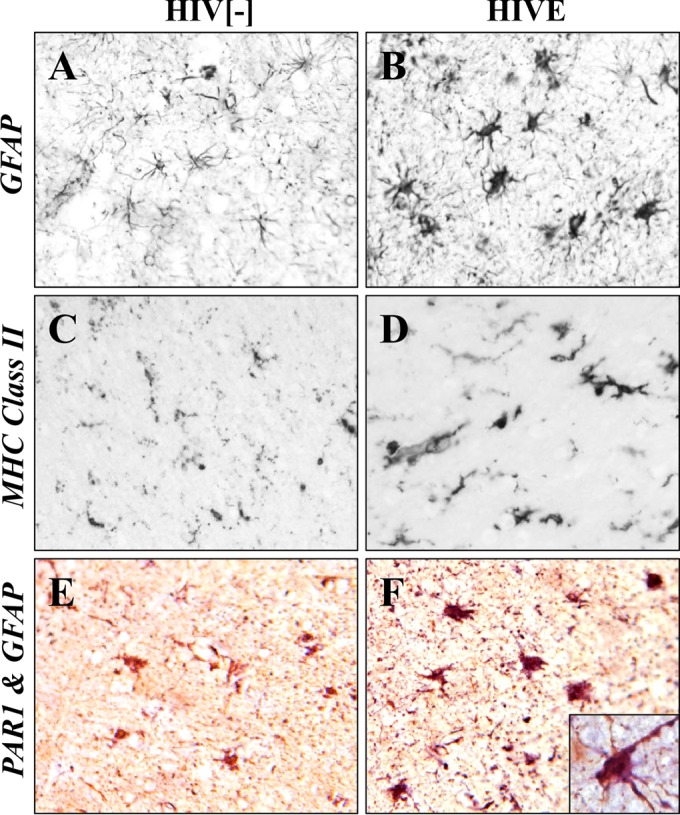
Increased PAR1 expression in astrocytes in HIVE. (A and B) GFAP immunoreactivity was infrequently detected in astrocytes in HIV− brain compared to results in brain from HIVE, which showed numerous hypertrophied GFAP-positive astrocytes. (C and D) Major histocompatibility antigen class II (MHC class II) immunoreactivity, a marker for activated microglia, was also increased in brain from HIVE compared to that in HIV− brain. (E) PAR1 immunoreactivity (purple) was minimally detected in GFAP-positive astrocytes (brown) in non-HIV brain. (F) PAR1 immunoreactivity (blue) was evident in GFAP-positive astrocytes in HIVE (purple results from blue plus brown), which was localized on the cell body and processes (inset). HIVE, HIV with encephalitis. Original magnification, ×20; inset magnification, ×40.
The deleterious effect of inflammatory cytokines in promoting immune response and inflammation via various mechanisms, including the generation of nitrogen intermediates, has been reported in cell lines such as pancreatic β cells, epithelial cells, and HeLa cells. Many of these studies show synergetic effects by utilizing a cocktail of cytokines instead of a single cytokine (45, 46). We adapted this method of using a cytokine cocktail that simulates inflammation in HeLa cells, which exhibited a robust response and expressed all four PCs (Fig. 1B). Thus, upon a 24-h exposure of HeLa cells to a mixture of TNF-α, IL-1β, and IFN-γ, the mRNA levels of furin, PC5, PACE4, and PC7 were upregulated by these inflammatory cytokines (2- to 4-fold) (Fig. 1B), independent of HIV infection. This was similarly observed only in HIV+ brains displaying encephalitis (Fig. 1A).
PAR1 and the PCs are coincidently expressed in mouse brain.
Upon analysis of the sequence of the N-terminal tethered ligand of PAR1 (NPR41SFLLR46↓]NP; possible cleavage sites in boldface), we hypothesized that PCs may cleave at Arg46↓ instead of its activating thrombin cleavage site Arg41↓, potentially leading to anti-inflammatory effects (23, 26). For this cleavage to occur, we hypothesized that PAR1 and enzymatically active PCs must be coexpressed in the same tissue and meet along the secretory pathway. To support our hypothesis, we first examined PAR1 and PC expression profiles in adult mouse brain to determine if the two proteins might be coexpressed in similar regions (Fig. 3). PAR1 is highly expressed in the olfactory lobe, hippocampus, dentate gyrus, and cerebellum. Furin and PC7 are also highly expressed in the olfactory lobe, hippocampus, cerebellum, and frontal cortex region. PC5 is rich in the cerebral cortex and dentate gyrus, while PACE4 is particularly abundant in the cerebellum. Notably, the frontal cortex and the hippocampus are major sites of motor and/or cognitive functions, which are selectively damaged in HAND (11). Although indicative of coexpression, this does not prove that the PAR1 and PC proteins are colocalized intracellularly. However, as presented later, we provide evidence for the protein cellular colocalization of PAR1 with membrane-bound PCs.
FIG 3.

Localization of PAR1 and PCs in adult mouse brain. In situ hybridization histochemistry was performed in cryosections of mouse brain using 35S-labeled cRNAs. Cb, cerebellum; Cx, cerebral cortex; DG, dentate gyrus; Hi, hippocampus; Hy, hypothalamus; Po, pons; OL, olfactory lobe; St, striatum.
The soluble PC5A and PACE4 cleave PAR1 at Arg46↓.
To examine PC cleavage of PAR1 directly, a synthetic peptide (19-mer) mimicking the N-terminal tethered ligand and putative PC cleavage site of human PAR1 was incubated with recombinant soluble furin, PC5A, or PACE4 or soluble PC7 (using equal enzymatic activities) to assess in vitro cleavage by these PCs, as we previously published (38). Digestion with trypsin, which cleaves the 19-mer at Arg41↓ (preferentially) and Arg46↓, was used as a reference for the resulting peptide fragments. By comparing the RP-HPLC profiles of the trypsin- and PC-digested peptide corresponding to human aa 35 to 53 of PAR1, it was determined that in vitro only PACE4 inefficiently (10% in 19 h) cleaves human PAR1 at the expected Arg46↓, separating most residues of the tethered ligand (SFLLR46↓N47) (Fig. 4).
FIG 4.
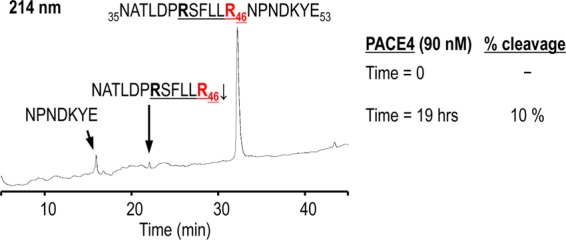
In vitro cleavage of PAR1 by PACE4. A synthetic peptide (19-mer) mimicking the tethered ligand and putative PC cleavage site of human PAR1 was used to assess in vitro cleavage by PCs. Comparative RP-HPLC analyses of PAR1 19-mer digestion with purified soluble PCs or trypsin (which cleaves preferentially at Arg41↓ and at Arg46↓) revealed that PACE4 cleaves human PAR1 at the expected Arg46↓. The products were separated by RP-HPLC on a Varian C18 column, and peptide bonds were detected at 214 nm. The percent cleavage was calculated as the ratio of the normalized peak area (peak area/number of peptide bonds) of C-terminal fragment NPNDKYE to that of the intact 19-mer peptide (at time zero).
To evaluate whether in cells such PC cleavage would be more efficient, we used a human PAR1 construct containing mCherry at its N terminus and eYFP at its C terminus (pcDNA3/mCherry-hPAR1-eYFP) (Fig. 5). Our published work evaluating the signaling properties and receptor dynamics of this dually tagged PAR1 construct indicates that the epitope-tagged receptor is fully functional, in keeping with the properties of the wild-type receptor (34). Accordingly, we coexpressed the mCherry-hPAR1-eYFP dually tagged PAR1 with and without furin, PC5A, PC5B, PACE4, or PC7 in HEK293 cells. We monitored the results of the coexpression of the PCs with the fluorescently tagged PAR1 utilizing confocal microscopy. The N-terminal ligand of PAR1 was intact when expressed alone as both N-terminal red and C-terminal yellow fluorescence signals were detected at the cell surface (Fig. 6, top panel). The cell surface expression of the PAR1 construct was confirmed by its colocalization with the low density lipoprotein receptor (LDLR), which is expressed and functions at the plasma membrane (Fig. 7) (47). However, LDLR does not necessarily colocalize with PAR1 in the endosomal-like structures near the cell surface (Fig. 7). Upon either short-term (e.g., 3 min) (34) or long-term (overnight) exposure of cells to thrombin (1 unit/ml), PAR1 lost its N-terminal red fluorescence and only a punctate C-terminal yellow fluorescence remained at the cell surface, confirming the thrombin cleavage and release of the N-terminal mCherry tag (Fig. 6, top row, Cherry-PAR1), as previously reported (23, 34). In addition, anti-thrombin III, which is a potent thrombin inhibitor, blocked the thrombin cleavage at Arg41 so that the N-terminal mCherry tag was retained (Fig. 6, middle row). Interestingly, coexpression of PAR1 with soluble PC5A or PACE4 removed the N-terminal red tag while preserving the C-terminal yellow tag, which was more evenly spread around the cell surface than that of thrombin-cleaved PAR1 (Fig. 6). While cleavage by thrombin at Arg41↓ does cause PAR1 to internalize (34) and results in a punctate pattern (Fig. 6), some enzymes, e.g., neutrophil elastase, can cleave PAR1 at a noncanonical site without causing receptor internalization (34). Our imaging data thus indicate that cleavage of PAR1 by PC5A and PACE4 results in a PAR1 form that loses its N terminus but does not internalize, resulting in a uniform distribution at the cell surface (Fig. 6). The PC cleavage site was confirmed at Arg46↓ since expression of these soluble PCs with a mutant PAR1 (R46A; P1 Ala) construct prevented the loss of the N-terminal mCherry fluorescent tag (Fig. 5 and 8). As the motif R41XXXXR46↓ in PAR1 is a general basic amino acid-specific PC motif (2), we also confirmed that the mutant R41A (P6 Ala) was not processed by either PC5A or PACE4 (data not shown). These data also demonstrated that PC5A and PACE4 cleavage of PAR1 does not occur within the mCherry domain since cleavage is prevented in the R46A mutant and since the sequence of mCherry does not contain a PC recognition motif. We determined that PAR1 cleavage occurs at the cell surface by demonstrating that it was completely blocked by two PC inhibitors (38): cell-permeable decanoyl-RVKR-cmk and nonpermeable D6R (Fig. 9). Finally, cleavage at Arg46↓ occurs only with soluble PCs as the membrane-bound PC5B and PC7 did not have any apparent effect on PAR1 (Fig. 10).
FIG 5.
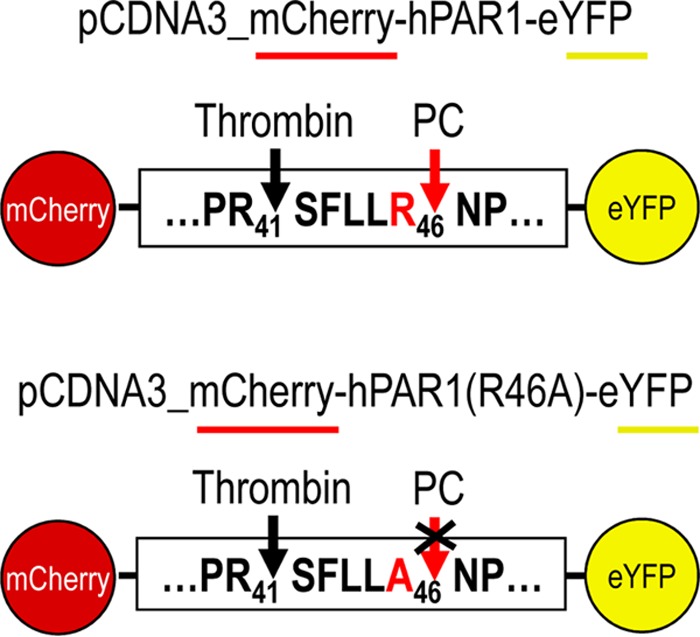
Schematic representation of human PAR1 constructs. Human PAR1 construct (wild-type and single-site mutant at Arg46) containing mCherry at its N terminus and eYFP at its C terminus was overexpressed with/without PCs in HEK293 cells to confirm the proteolytic cleavage of PAR1 by PCs. While noncleaved PAR1 would contain both mCherry (red) and eYFP (yellow) fluorescence, cleaved-PAR1 would lose mCherry fluorescence.
FIG 6.

Ex vivo cleavage of PAR1 by soluble PC5A and PACE4. A human PAR1 construct containing mCherry at its N terminus and eYFP at its C terminus was overexpressed with PCs in HEK293. Thrombin cleaves PAR1 at Arg41↓, and its activity is inhibited by anti-thrombin III. Soluble PC5A and PACE4 cleave PAR1 at Arg46↓. Scale bars, 20 μm.
FIG 7.

PAR1 colocalizes with the cell surface low-density lipoprotein receptor (LDLR). A human PAR1 construct containing mCherry at its N terminus and eYFP at its C terminus overexpressed with LDLR-V5 (green; detected with an Alexa Fluor 647-conjugated secondary donkey anti-mouse antibody) in HEK293 demonstrates the colocalization of PAR1 and LDLR at the cell surface. Scale bar, 20 μm.
FIG 8.

Cleavage of PAR1 at Arg46 by PC5A and PACE4. A single-amino acid variant of human PAR1 at Arg46 was coexpressed with PC5A and PACE4. The mutant prevented the cleavage, indicating that PC5A and PACE4 cleave at Arg46. Scale bars, 20 μm.
FIG 9.

Cleavage of PAR1 by PC5A and PACE4 on cell surface. Both PC specific inhibitors, cell-permeable RVKR-cmk and nonpermeable D6R, prevented the cleavage of PAR1, indicating that PC5A and PACE4 cleave PAR1 on cell surface rather than in the TGN. Scale bars, 20 μm.
FIG 10.

No effect of membrane-bound PC5B and PC7 on PAR1. A human PAR1 construct containing mCherry at its N terminus and eYFP at its C terminus was overexpressed with PCs in HEK293. Exogenous overnight addition of 1 unit/ml thrombin cleaved PAR1 while membrane-bound PC5B and PC7 had no effect. Scale bars, 20 μm.
PACE4 and PC5A reduce calcium mobilization in response to thrombin stimulation.
PAR1 activation by thrombin signals via G proteins and leads to Gαq-mediated calcium release, which ultimately results in inflammation (21). Thrombin cleavage of PAR1 has been previously demonstrated to induce calcium mobilization (34). In order to evaluate the inhibitory effects of PACE4 and PC5A on thrombin-induced calcium mobilization, we transfected HEK293 cells, which endogenously express PAR1, with cDNAs coding for PACE4 or PC5A and then monitored their effect on thrombin-induced calcium mobilization (Fig. 11). The data showed that PACE4 and, less so, PC5A significantly reduced by ∼2.5-fold the amount of calcium mobilization in response to thrombin stimulation. This result agrees with the hypothesis that PACE4 and PC5A would cleave PAR1 at Arg46, thereby disarming PAR1 for thrombin activation and attenuating the calcium mobilization induced by thrombin cleavage of PAR1 at Arg41. Notably, since PCs require both P1 Arg46 and P6 Arg41 for substrate recognition, the reduced thrombin-induced calcium signaling in cells overexpressing PACE4 or PC5A means that the PCs must have cleaved PAR1 at the cell surface before thrombin, which would have otherwise removed the P6 Arg41 and prevented PC cleavage. However, we cannot exclude the possibility that proteins in addition to PAR1 in HEK293 cells are also sensitive to PACE4 and PC5A so as to diminish thrombin-induced calcium mobilization via a concurrent non-PAR1 mechanism.
FIG 11.

PACE4 and PC5A cleavage of PAR1 decreases calcium mobilization in response to thrombin stimulation. HEK293 cells were mock transfected or transiently transfected with cDNAs coding for PACE4 or PC5A, as indicated. The next day the cells were incubated for 30 min with fluo-4-AM no-wash calcium indicator dye and then treated with 20 nM thrombin. Intracellular calcium mobilization was immediately assayed by the fluorescence signal (see Materials and Methods). Representative plots of the nonlinear fits of relative fluorescence measured at 516 nm are shown in panel A, and the peak heights are compared in panel B (n = 3). ⭑, P < 0.05, one-way analysis of variance with Dunnett's post hoc test.
Furin causes PAR1 retention in the TGN.
Unlike the membrane-bound PC5B and PC7 (Fig. 10), furin relocalizes PAR1 to the TGN (Fig. 12, colocalized with the golgin-97 marker). Additionally, furin does not seem to cleave PAR1 as both red and yellow fluorescence were detected in the TGN. In contrast, overexpression of the PAR1 R46A mutant did not result in PAR1 relocalization to the TGN, indicating that this effect requires furin to bind to the N-terminal domain of PAR1 in the Arg46 region (Fig. 12). Furthermore, the mutant R41A (P6 Ala) or the double mutant R41A R46A (P6 and P1 Ala) does not affect the subcellular localization of PAR1 in the presence of furin (data not shown). These data argue for the binding of PAR1 at the active site of furin, which likely recognizes the R41XXXXR46 sequence (2) of PAR1 but does not cleave the receptor.
FIG 12.

PAR1 retention in the TGN by furin. A human PAR1 construct containing mCherry at its N terminus and eYFP at its C terminus overexpressed with furin in HEK293 results in PAR1 retention in the trans-Golgi network as confirmed by a TGN marker, golgin-97. Scale bars, 20 μm.
We next sought to define the mechanism behind this relocalization of PAR1 to the TGN in the presence of furin and to determine if the phospho-furin acidic cluster sorting protein 1 (PACS1) regulates this process. PACS1 has been reported to bind the serine-phosphorylated acidic domain of the cytosolic tail (CT) of furin (EECPS773DS775EEDE; serines are underlined), thereby redirecting the localization of furin to the TGN (48). Therefore, it was possible that via PACS1 binding to the CT of furin, the furin-PAR1 complex would be redirected to the TGN. Furthermore, since it was reported that the CT of PAR1 is phosphorylated because it contains an acidic serine-rich sequence, ESSDPSSYNSSGQLMAS406 (49), we speculated that, like furin, PAR1 could also be redirected to the TGN in the presence of excess PACS1. In support of this hypothesis, a relocalization of PAR1 to the TGN was also observed upon coexpression of PAR1 with PACS1 (Fig. 13).
FIG 13.
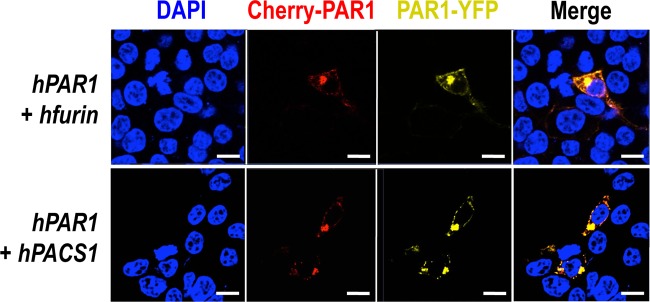
PAR1 retention in the TGN by PACS1. A human PAR1 construct containing mCherry at its N terminus and eYFP at its C terminus overexpressed with PACS1, a transporting protein of furin from early endosome to TGN, results in similar PAR1 retention in the TGN. Scale bars, 20 μm.
PAR1 reduces cleavage activity of the membrane-bound furin and PC5B, partially inhibits PC7 activity, and colocalizes with furin and PC7.
HIV glycoprotein gp160 is cleaved by furin to produce gp120 and gp41 (Fig. 14A and B), which are critical components for viral entry and infection of host cells (28–30). In COS-1 cells, cleavage of gp160 by endogenous convertases or by overexpressed furin (38) into gp120 was markedly reduced (40 to 50%) in the presence of PAR1 (Fig. 14A). PAR1 R46A does not inhibit the gp160 processing by furin (Fig. 14B), indicating that Arg46 plays a critical role in the inhibitory effect of PAR1, likely through Arg46 binding to the catalytic domain of furin.
FIG 14.

Inhibition of furin cleavage of HIV glycoprotein gp160 by PAR1. Human PAR1 was transiently overexpressed with PCs in COS-1 cells. (A) PAR1 inhibits furin processing of gp160 to gp41. (B) A PAR1 mutant (R46A) prevents inhibition on furin processing. (C) PAR1 does not block processing of Vpr by soluble PACE4. WB, Western blotting. FP, fusion peptide; M, membrane.
The HIV accessory protein Vpr, which has neurotoxic properties, is cleaved and inactivated by soluble PC5A and PACE4 (10) (Fig. 14C). However, and in accordance with the ability of these convertases to process PAR1 (Fig. 6), this cleavage is not inhibited by PAR1 (Fig. 14C).
It was previously shown that PC5A and PC5B can similarly process the proform of GDF11 (proGDF11) into GDF11 (37). As evidence for a differential effect of PAR1 on the two PC5 isoforms, we demonstrate that the PC5A processing of proGDF11 is not affected, while cleavage by PC5B is blocked by PAR1 (Fig. 15A). This result reveals that PAR1 is an inhibitor of PC5B but not PC5A.
FIG 15.

PAR1 inhibition of membrane-bound PC5B and PC7. Human PAR1 was transiently overexpressed with PC5A, PC5B, or PC7 in HEK293 cells. (A) PAR1 inhibits processing of proGDF11 by PC5B, not PC5A. The structures of PC5A and PC5B are illustrated below. The arrows represent the autocatalytic cleavage sites. The positions of the active site Asp, His, and Ser and the oxyanion hole Asn are also shown. The transmembrane domain (gray) of PC5B is before the green cytosolic tail. The Cys-rich domain is represented in pink. (B) PAR1 partially inhibits PC7 processing of human TfR1. FG, Flag; s-hTfR1, soluble human TfR1.
In HEK293 cells the PC7-specific cleavage of human TfR1 (5) is only partially (∼25 to 30%) inhibited by PAR1 (Fig. 15B), likely due to the fact that the steady-state localization of activated PC7 is in punctate-like structures just below the cell surface (6). This subcellular location may not be readily accessible to PAR1. We conclude that PAR1 inhibits the membrane-bound furin, PC5B, and partially PC7 but is cleaved by soluble PC5A and PACE4.
In order to monitor the colocalization of the membrane-bound PCs and PAR1 on a more cellular level, we employed a Duolink in situ proximity ligation assay (PLA). The PLA detects the physical proximity of two proteins by utilizing primary antibodies from two different species, such as rabbit and mouse, against each protein of interest. The secondary PLA probes (anti-rabbit antibody plus sense and anti-mouse antibody minus sense) interact with their respective primary antibodies, and if within close proximity this interaction will lead to the production of PCR-amplified distinct fluorescent dots that can be quantitated by fluorescence microscopy. Since PC5B is expressed only in the small intestine and adrenal cortex (50) while furin and PC7 are abundant in most tissues, including brain (2), we sought to analyze the colocalization of PAR1 with furin and PC7. The data clearly show that both enzymes interact with mCherry-hPAR1-eYFP when cotransfected into HEK293 cells, in contrast to results with cells cotransfected with mCherry-hPAR1-eYFP and an empty pIRES vector (Fig. 16). Thus, based on the distribution of the number of dots/cell, we estimate that on average cells ini which PAR1 colocalizes with furin, PC7, or an empty vector show 35, 23, or <2 dots/cell, respectively. This result affirms the close intracellular proximity of PAR1 with the membrane-bound furin and PC7. This interaction very likely explains the inhibitory effect of PAR1 on the proteolytic activities of furin and PC7 and the cellular relocalization of PAR1 by furin.
FIG 16.

A proximity ligation assay (PLA) demonstrates the interaction of furin and PC7 with PAR1. HEK293 cells were transfected with cDNAs as indicated and incubated with rabbit anti-PAR1 and mouse anti-V5 primary antibodies. PLA probes, anti-rabbit antibody minus sense and anti-mouse antibody plus sense, were utilized to detect the interaction of mCherry-hPAR1-eYFP with V5-tagged furin or PC7. The dots (indicating interactions of PLA probes) per cell were manually counted. The enlarged pictures emphasize transfected cells where PAR1 colocalizes with either furin or PC7, indicated by green dots. Based on the distribution of dots/cell (right panels), we determined that control cells averaged <2 dots/cell whereas those that express furin or PC7 averaged 35 and 23 dots/cell, respectively. Scale bars, 15 μm.
PAR1 inhibits the processing of HIV-1 gp160 and significantly reduces viral infectivity.
Having demonstrated that PAR1 affects the processing of gp160 into gp120 and gp41 (Fig. 14A and B), we next set out to determine if this inhibition of gp160 processing had a direct impact on virus infectivity. Accordingly, HEK293T cells were cotransfected with the NL4-3 HIV-1 proviral plasmid (40) and different amounts of cDNAs coding for either mCherry-hPAR1-eYFP or its R46A mutant. Following 2 days of cell culture, the cell lysates were analyzed by Western blotting using a panel of antibodies against p24, gp160/gp120, gp41, and GAPDH, as well as GFP as a measure of mCherry-hPAR1-eYFP protein. As previously observed in COS-1 cells (Fig. 14A and B), gp160 processing was also inhibited by overexpression of PAR1 in HEK293T cells but not by that of its R46A mutant (Fig. 17A and B).
FIG 18.
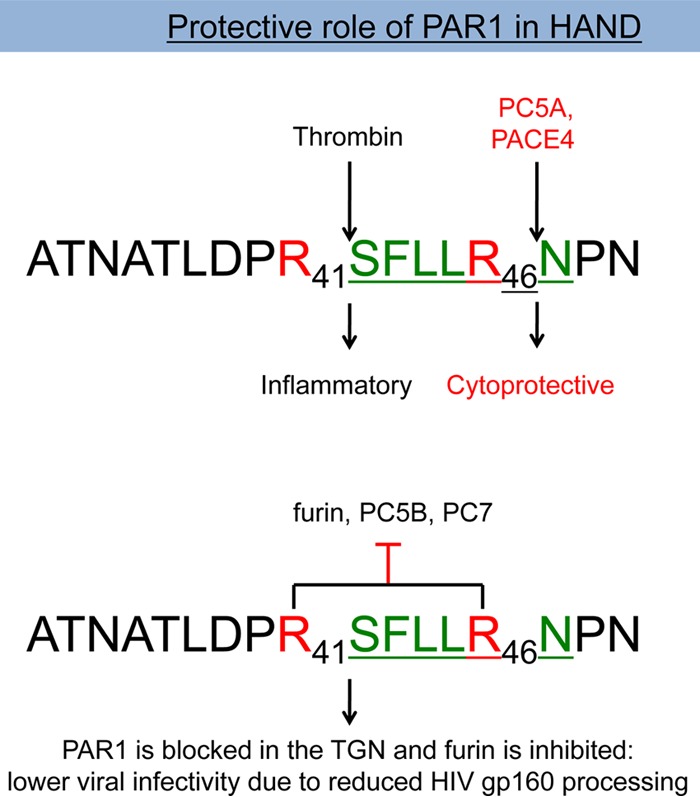
Schematic diagram of the effects of PAR1 on PCs. Human PAR1 is processed by thrombin at Arg41↓ and by the soluble PC5A and PACE4 at Arg46↓, resulting in inflammatory and cytoprotective effects, respectively. On the other hand, PAR1 inhibits the membrane-bound furin and PC5B and partially PC7 and is relocalized to the TGN by furin.
FIG 17.

Inhibition of furin processing of gp160 of HIV-1 by PAR1 reduces virus infectivity. (A) HEK293T cells were cotransfected with the indicated amounts of plasmids (μg), and cellular protein expression was analyzed following SDS-PAGE and Western blotting, as described in Materials and Methods. (B) Quantitative analyses of selected intracellular viral proteins. (C) Infection assay for HIV-1 viruses. HEK293T cells were transfected with 5 μg of proviral NL4-3 DNA together with either 0.1 μg of mCherry-hPAR1-eYFP R46A mutant plus 0.9 μg of control EGFP-C1 vector or with 0.1 μg of WT mCherry-hPAR1-eYFP plus 0.9 μg of control EGFP-C1 vector. A dilution curve of viral load confirmed nonsaturating conditions (1 ng of virus per 750,000 TZM-bl cells). Note the ∼70% inhibition of virus infectivity by WT PAR1. ***, P < 0.001 (Student's t test). These data are representative of at least two independent experiments.
In order to further quantify the impact of PAR1 overexpression on HIV-1, we measured the infectivity of viral particles produced from cotransfected HEK293T cells with 0.1 μg of cDNAs coding for PAR1 or PAR1 R46A using the TZM-bl reporter cell line (43). Three viral loads (1, 0.2, and 0.04 ng of p24) were tested, and the 0.04-ng virus load was found to be nonsaturating in the assay parameters (data not shown). Under these viral load conditions, we observed a significant (P = 3 × 10−13) 3-fold decrease in infectivity of viruses obtained from HEK293T cells overexpressing PAR1 compared to infectivity of the R46A mutant (Fig. 17C). Thus, PAR1 overexpression inhibits the infectious potential of released HIV-1 viral particles. These data represent the first evidence that PAR1 can reduce HIV-1 viral infectivity, likely due to its inhibition of viral glycoprotein processing by furin.
DISCUSSION
Higher transcript and protein levels of PAR1, a proinflammatory thrombin-cleaved GPCR, have previously been associated with HIV encephalitis (22), a result also observed in our HAND cohort with encephalitis (Fig. 1A). Cleavage of PAR1 by thrombin at Arg41↓ exposes an internal N-terminal sequence, SFLLR46N, that is believed to bind to the second extracellular loop of PAR1 via an Arg46/Glu260 ionic interaction, resulting in a proinflammatory response and an increase in calcium mobilization. However, it has also been demonstrated that PAR1 is cleaved by neutrophil elastase (34) and APC (26) at SFLL↓R46N and SFLLR46↓N, respectively. This cleavage disarms the tethered ligand SFLLR46N, to prevent PAR1 activation by thrombin, and instead generates a biased PAR1 tethered ligand signal that leads to an anti-inflammatory/cytoprotective response (24–26, 34).
PCs can activate metalloproteinases that are responsible for the generation of inflammatory molecules such as TNF-α (2) or for protection against inflammation via production of transforming growth factor β (TGF-β) (51). However, a link between the PCs and the direct processing of a GPCR that mediates inflammation has not been reported. Herein, we report that in brains from patients with HAND and neuroinflammation, verified by the presence of encephalitis (Fig. 1A), there is a marked increased expression of PAR1 and of four closely related basic amino acid-specific PCs: furin, PC5, PACE4, and PC7 (2) (Fig. 1A). The upregulation of these PCs was also observed in HeLa cells treated with a cocktail of inflammatory cytokines (Fig. 1B). The collective upregulation PAR1 and PCs in HAND brains suggests a possible role of the PCs in neuroinflammation and neurodegeneration.
Our studies provide evidence that PAR1 is cleaved at Arg46↓ by the soluble convertases PC5A and PACE4 (Fig. 6 and 8), likely disarming the PAR1 tethered ligand, SFLLR46N, and reducing thrombin's effect on PAR1. In support of this conclusion, we also observed that overexpression of either PACE4 or PC5A in HEK293 cells expressing PAR1 endogenously results in a significant decrease in calcium mobilization in response to thrombin (Fig. 11). Additionally, it is expected that processing of PAR1 by either PACE4 or PC5A primarily takes place at the cell surface, in accordance with the location of the zymogen activation of these convertases, which occurs via binding to cell surface HSPGs (4). This conclusion is supported by the lack of cleavage of PAR1 by these PCs in the presence of heparin, which prevents their binding to HSPGs and hence their zymogen activation (4) (data not shown).
Interestingly, proGDF11 is cleaved by PC5A/B and PACE4 only at the sequence RSRR296↓N, which exhibits a critical Asn297 (underlined) at the P1′ site. Replacement of this Asn297 by Asp297 resulted in the ability of all convertases to process proGDF11 (37). Coincidentally, in mammals this highly conserved P1′ Asn47 follows the PAR1 cleavage site SFLLR46↓N, which might rationalize its selective processing by PC5A and PACE4. Further investigation is required to determine if mutation of Asn47 to Asp47 in PAR1 would also lead to the lack of selectively in its processing by PCs, as for proGDF11, and possibly the loss of furin inhibition.
While PC5A, PACE4, and APC cleave cell surface PAR1 at Arg46↓, APC does so only at the surface of endothelial cells. In these cells, the thrombomodulin-mediated binding of thrombin greatly enhances its ability to activate the anticoagulant protein C into APC on the vascular wall (25). Furthermore, binding of protein C to its endothelial receptor (EPCR, for endothelial protein C receptor) is required for its effective activation by thrombin via its cleavage at ProArg221↓ yielding APC (52). This protease in turn cleaves PAR1 at Arg46↓ (24). The resulting PAR1 cleaved at Arg46↓ is reported to promote cytoprotective signaling via a noncanonical PAR1 tethered ligand (24–26). However, in the brain APC mRNA is expressed at low levels (53). In contrast, PC5 (50) and PAR1 are widely expressed in the brain, while PACE4 (54) and PAR1 are rich in the cerebellum (Fig. 3). Our colocalization data support the notion that in the brain both PC5A and PACE4 could process endogenous PAR1, unmasking the N47-tethered ligand, leading to an anti-inflammatory response. This process may represent a protective reaction to the observed inflammation in HAND patients with encephalitis (Fig. 1 and 2).
Recently, we reported that PAR1 can inhibit furin processing of the Fc-viral glycoprotein of human metapneumovirus by an unknown mechanism (27). In the present study, we demonstrated that PAR1 can inhibit the proteinase activity of the three membrane-bound basic amino acid-specific PCs (2), namely, furin on HIV gp160, PC5B on proGDF11, and partially PC7 on human TfR1 (Fig. 14A and 15). While the PC5B isoform is predominantly expressed in the small intestine and adrenal cortex (50), the expression of furin and PC7 is widespread and enriched in the brain (Fig. 3). We chose HIV gp160 as a relevant substrate for furin (28), Vpr for PACE4 (10), GDF11 for PC5A and PC5B (37), and human TfR1 as a PC7-specific substrate (5). The data clearly demonstrate that, for the four selected substrates, the expression of PAR1 is associated with a loss of cleavage activity of membrane-bound convertases but not soluble ones and that the PAR1 mutants R46A or R41A R46A are not inhibitory. This result established that inhibition of the PC activity requires the recognition of the R41XXXXR46 motif in PAR1 by the catalytic subunit of the membrane-bound basic amino acid-specific PCs (Fig. 18). We envisage that PAR1 binds these membrane-bound PCs via this motif, without cleavage occurring, likely due in part to the presence of the Asn47 at the P1′ position of PAR1. However, it is not clear why the membrane-bound PCs are inhibited and not the soluble ones, especially since PAR1 inhibits proGDF11 cleavage by PC5B but not by PC5A (Fig. 15A). Thus, it appears that membrane attachment of PCs is a critical factor for the ability of the seven-transmembrane GPCR PAR1 to inhibit them, likely due to an optimal proximity effect. Indeed, using a proximity ligation assay, we demonstrated that furin and, less so, PC7 interact closely with PAR1 (Fig. 16), thereby conferring the PAR1 inhibitory effect on these PCs (Fig. 14A and 15B).
Our data demonstrated that while PAR1 inhibits the protease activity of PC5B (Fig. 15A) but only partially that of PC7 (Fig. 15B), the localization of PAR1 at the cell surface colocalizing with the LDLR (Fig. 7) is unaffected by these proteases (Fig. 10). However, while PAR1 inhibits furin activity, overexpression of furin results in a predominant concentration of wild-type PAR1 but not its R46A mutant in the TGN (Fig. 12). The TGN is a region where the steady-state concentration of furin is highest (3). Since binding of the cytosolic adapter protein PACS1 to the serine-phosphorylated CT of furin results in its recycling from the cell surface to the TGN (3), it was plausible that the phospho-CT-furin–PAR1 complex could also be recruited to the TGN via PACS1. Indeed, overexpression of either PACS1 or furin resulted in a similar TGN recruitment of PAR1 (Fig. 13). Whether endogenous furin in HEK293 cells is enough to explain the PACS1-induced relocalization of PAR1 to the TGN or whether overexpressed PACS1 would also bind directly to the CT of PAR1 that has been reported to be serine phosphorylated (49) is not yet clear. Mutagenesis of the acidic serine-rich sequence ESSDPSSYNSSGQLMAS406 in the CT of PAR1 may help to elucidate this process. However, the fact that endogenous furin/PACS1 in HEK293 cells did not result in an enrichment of overexpressed PAR1 in the TGN (Fig. 6) indicates that the level of endogenous furin-PACS1 complex may be limiting and that the overexpression of either partner may be enough to relocalize PAR1 to the TGN. Since a stoichiometry of 1:1 would be expected for the furin-PAR1 complex (Fig. 16) and since PACS1 is only transiently used and reused to sort these proteins to the TGN, it is more likely that endogenous furin may be the limiting factor in this process. Accordingly, overexpression of PAR1 versus PAR1-furin in PACS1-depleted cells would also be informative.
Furin is one of the major PCs that process the HIV-1 gp160 into gp120/gp41 (Fig. 14A) to allow viral fusion and entry into cells (28, 29). In addition, a screen of a small interfering RNA library directed against 5,000 genes identified furin as one of the potential therapeutic targets implicated in HIV-1 infection (55). Interestingly, and of relevance to HAND, HIV can be detected in the frontal cortex soon after primary infection (11), a region of the brain rich in furin expression (Fig. 3). Thus, even though inflammation increases the levels of both furin and PAR1 in the brain (Fig. 1), the latter would inhibit furin, limiting its ability to process gp160, and hence could play a protective role to dampen viral infection. Our data support this model as overexpression of PAR1, but not that of its R46A mutant that does not inhibit furin (Fig. 14A and B), resulted in a 3-fold reduction in HIV-1 cellular infectivity (Fig. 17C). In this context, we would like to emphasize that PAR1 represents the first identified endogenous protein that selectively inhibits the activity of membrane-bound convertases, including furin. Identification of the mechanistic details of such inhibition and the ability of furin to relocalize PAR1 to the TGN will surely enhance our understanding of the relevance of PAR1 to HAND. Additionally, the possibility that human PAR2 and PAR3, which also exhibit the PC-like motifs (2) RSSKGR36 and KTFR41, respectively, are regulated by PCs will need to be further investigated. Finally, the increased PAR1 expression in cardiovascular/immune/nervous systems, inflammation, and cancer (56) may also inhibit furin in these pathologies.
In conclusion, the brains from HAND patients in association with neuroinflammation exhibit increased expression of PAR1 and four basic amino acid-specific PCs. Our results suggest that the inflammation-induced upregulation of PAR1 and the PCs might be neuroprotective. On one hand, PC5A and PACE4 disarm PAR1, while, on the other hand, PAR1 inhibits furin and PC7, which are the major enzymes that cleave gp160 into gp120/gp41 (28), likely ensuring that viral infectivity is diminished (Fig. 17C). Thus, modulation of PC activity might represent a potential therapeutic approach to limit the onset and progression of neuroinflammation and neurodegeneration in persons with HAND.
ACKNOWLEDGMENTS
This work was supported by a Canadian Institutes of Health Research (CIHR) Emerging Team Grant on HAND 267359 (N.G.S., C.P., and E.A.C.) and in part by a CIHR operating grant (M.D.H.). N.G.S., C.P., and E.A.C. hold Canada Research Chairs in Precursor Proteolysis, Neurological Infection and Immunity, and Human Retrovirology, respectively. W.K. was a recipient of an IRCM Bourse de Doctorat Jacques Gauthier. B.G. was supported by U24MH100930.
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Chessie 13-39.1 and Chessie 8 from George Lewis, hybridoma 902 (anti-gp120) from Bruce Chesebro, plasmid NL4-3 from Malcolm Martin, and TZMbl from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc. We thank M. Bego for help with the PLA, W. Branton and J. Hamelin for technical assistance, and Brigitte Mary for secretarial help.
REFERENCES
- 1.Seidah NG, Mayer G, Zaid A, Rousselet E, Nassoury N, Poirier S, Essalmani R, Prat A. 2008. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol 40:1111–1125. doi: 10.1016/j.biocel.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 2.Seidah NG, Prat A. 2012. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov 11:367–383. doi: 10.1038/nrd3699. [DOI] [PubMed] [Google Scholar]
- 3.Thomas G. 2002. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer G, Hamelin J, Asselin MC, Pasquato A, Marcinkiewicz E, Tang M, Tabibzadeh S, Seidah NG. 2008. The regulated cell surface zymogen activation of the proprotein convertase PC5A directs the processing of its secretory substrates. J Biol Chem 283:2373–2384. doi: 10.1074/jbc.M708763200. [DOI] [PubMed] [Google Scholar]
- 5.Guillemot J, Canuel M, Essalmani R, Prat A, Seidah NG. 2013. Implication of the proprotein convertases in iron homeostasis: proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology 57:2514–2524. doi: 10.1002/hep.26297. [DOI] [PubMed] [Google Scholar]
- 6.Rousselet E, Benjannet S, Hamelin J, Canuel M, Seidah NG. 2011. The proprotein convertase PC7: unique zymogen activation and trafficking pathways. J Biol Chem 286:2728–2738. doi: 10.1074/jbc.M110.192344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pasquato A, Palma JR, Galan C, Seidah NG, Kunz S. 2013. Viral envelope glycoprotein processing by proprotein convertases. Antiviral Res 99:49–60. doi: 10.1016/j.antiviral.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Garten W, Hallenberger S, Ortmann D, Schafer W, Vey M, Angliker H, Shaw E, Klenk HD. 1994. Processing of viral glycoproteins by the subtilisin-like endoprotease furin and its inhibition by specific peptidylchloroalkylketones. Biochimie 76:217–225. doi: 10.1016/0300-9084(94)90149-X. [DOI] [PubMed] [Google Scholar]
- 9.Vollenweider F, Benjannet S, Decroly E, Savaria D, Lazure C, Thomas G, Chretien M, Seidah NG. 1996. Comparative cellular processing of the human immunodeficiency virus (HIV-1) envelope glycoprotein gp160 by the mammalian subtilisin/kexin-like convertases. Biochem J 314:521–532. doi: 10.1042/bj3140521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao Y, Chen G, Richard J, Rougeau N, Li H, Seidah NG, Cohen EA. 2008. Cell-surface processing of extracellular human immunodeficiency virus type 1 Vpr by proprotein convertases. Virology 372:384–397. doi: 10.1016/j.virol.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ragin AB, Wu Y, Gao Y, Keating S, Du H, Sammet C, Kettering CS, Epstein LG. 2015. Brain alterations within the first 100 days of HIV infection. Ann Clin Transl Neurol 2:12–21. doi: 10.1002/acn3.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nath A. 2015. Eradication of human immunodeficiency virus from brain reservoirs. J Neurovirol 21:227–234. doi: 10.1007/s13365-014-0291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. 2007. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clifford DB, Ances BM. 2013. HIV-associated neurocognitive disorder. Lancet Infect Dis 13:976–986. doi: 10.1016/S1473-3099(13)70269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen MF, Gill AJ, Kolson DL. 2014. Neuropathogenesis of HIV-associated neurocognitive disorders: roles for immune activation, HIV blipping and viral tropism. Curr Opin HIV AIDS 9:559–564. doi: 10.1097/COH.0000000000000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Power C, Hui E, Vivithanaporn P, Acharjee S, Polyak M. 2012. Delineating HIV-associated neurocognitive disorders using transgenic models: the neuropathogenic actions of Vpr. J Neuroimmune Pharmacol 7:319–331. doi: 10.1007/s11481-011-9310-7. [DOI] [PubMed] [Google Scholar]
- 17.Iskander S, Walsh KA, Hammond RR. 2004. Human CNS cultures exposed to HIV-1 gp120 reproduce dendritic injuries of HIV-1-associated dementia. J Neuroinflammation 1:7. doi: 10.1186/1742-2094-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vivithanaporn P, Heo G, Gamble J, Krentz HB, Hoke A, Gill MJ, Power C. 2010. Neurologic disease burden in treated HIV/AIDS predicts survival: a population-based study. Neurology 75:1150–1158. doi: 10.1212/WNL.0b013e3181f4d5bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, Masliah E, Commins DL, Brandt D, Grant I, Singer EJ, Levine AJ, Miller J, Winkler JM, Fox HS, Luxon BA, Morgello S. 2012. The National NeuroAIDS Tissue Consortium brain gene array: two types of HIV-associated neurocognitive impairment. PLoS One 7:e46178. doi: 10.1371/journal.pone.0046178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noorbakhsh F, Vergnolle N, Hollenberg MD, Power C. 2003. Proteinase-activated receptors in the nervous system. Nat Rev Neurosci 4:981–990. doi: 10.1038/nrn1255. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. 2012. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov 11:69–86. doi: 10.1038/nrd3615. [DOI] [PubMed] [Google Scholar]
- 22.Boven LA, Vergnolle N, Henry SD, Silva C, Imai Y, Holden J, Warren K, Hollenberg MD, Power C. 2003. Up-regulation of proteinase-activated receptor 1 expression in astrocytes during HIV encephalitis. J Immunol 170:2638–2646. doi: 10.4049/jimmunol.170.5.2638. [DOI] [PubMed] [Google Scholar]
- 23.Ishii K, Gerszten R, Zheng YW, Welsh JB, Turck CW, Coughlin SR. 1995. Determinants of thrombin receptor cleavage. Receptor domains involved, specificity, and role of the P3 aspartate. J Biol Chem 270:16435–16440. [DOI] [PubMed] [Google Scholar]
- 24.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. 2012. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 120:5237–5246. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mosnier LO, Zlokovic BV, Griffin JH. 2007. The cytoprotective protein C pathway. Blood 109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 26.Schuepbach RA, Madon J, Ender M, Galli P, Riewald M. 2012. Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost 10:1675–1684. doi: 10.1111/j.1538-7836.2012.04825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aerts L, Hamelin M-E, Rhéaume C, Lavigne S, Couture C, Kim W, Susan-Resiga D, Prat A, Seidah NG, Vergnolle N, Boivin G. 2013. Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. PLoS One 8:e72529. doi: 10.1371/journal.pone.0072529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Decroly E, Wouters S, Di Bello C, Lazure C, Ruysschaert JM, Seidah NG. 1996. Identification of the paired basic convertases implicated in HIV gp160 processing based on in vitro assays and expression in CD4+ cell lines. J Biol Chem 271:30442–30450. doi: 10.1074/jbc.271.48.30442. [DOI] [PubMed] [Google Scholar]
- 29.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360:358–361. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 30.Moulard M, Decroly E. 2000. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim Biophys Acta 1469:121–132. doi: 10.1016/S0304-4157(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 31.Noorbakhsh F, Tsutsui S, Vergnolle N, Boven LA, Shariat N, Vodjgani M, Warren KG, Andrade-Gordon P, Hollenberg MD, Power C. 2006. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med 203:425–435. doi: 10.1084/jem.20052148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, Broadhurst DI, Power C. 2014. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 11:35. doi: 10.1186/1742-4690-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. 2003. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mihara K, Ramachandran R, Renaux B, Saifeddine M, Hollenberg MD. 2013. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J Biol Chem 288:32979–32990. doi: 10.1074/jbc.M113.483123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozden S, Lucas-Hourani M, Ceccaldi PE, Basak A, Valentine M, Benjannet S, Hamelin J, Jacob Y, Mamchaoui K, Mouly V, Despres P, Gessain A, Butler-Browne G, Chretien M, Tangy F, Vidalain PO, Seidah NG. 2008. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J Biol Chem 283:21899–21908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- 36.Lodge R, Lalonde JP, Lemay G, Cohen EA. 1997. The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J 16:695–705. doi: 10.1093/emboj/16.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Essalmani R, Zaid A, Marcinkiewicz J, Chamberland A, Pasquato A, Seidah NG, Prat A. 2008. In vivo functions of the proprotein convertase PC5/6 during mouse development: Gdf11 is a likely substrate. Proc Natl Acad Sci U S A 105:5750–5755. doi: 10.1073/pnas.0709428105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Susan-Resiga D, Essalmani R, Hamelin J, Asselin MC, Benjannet S, Chamberland A, Day R, Szumska D, Constam D, Bhattacharya S, Prat A, Seidah NG. 2011. Furin is the major processing enzyme of the cardiac-specific growth factor bone morphogenetic protein 10. J Biol Chem 286:22785–22794. doi: 10.1074/jbc.M111.233577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abacioglu YH, Fouts TR, Laman JD, Claassen E, Pincus SH, Moore JP, Roby CA, Kamin-Lewis R, Lewis GK. 1994. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res Hum Retroviruses 10:371–381. doi: 10.1089/aid.1994.10.371. [DOI] [PubMed] [Google Scholar]
- 40.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chesebro B, Wehrly K. 1988. Development of a sensitive quantitative focal assay for human immunodeficiency virus infectivity. J Virol 62:3779–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pincus SH, Wehrly K, Chesebro B. 1989. Treatment of HIV tissue culture infection with monoclonal antibody-ricin A chain conjugates. J Immunol 142:3070–3075. [PubMed] [Google Scholar]
- 43.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi Y, McClure MO, Pizzato M. 2008. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J Virol 82:12585–12588. doi: 10.1128/JVI.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Babcock TA, Carlin JM. 2000. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine 12:588–594. doi: 10.1006/cyto.1999.0661. [DOI] [PubMed] [Google Scholar]
- 46.Cetkovic-Cvrlje M, Eizirik DL. 1994. TNF-alpha and IFN-gamma potentiate the deleterious effects of IL-1 beta on mouse pancreatic islets mainly via generation of nitric oxide. Cytokine 6:399–406. doi: 10.1016/1043-4666(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 47.Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chretien M, Prat A, Seidah NG. 2004. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 48.Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, Thomas G. 1998. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94:205–216. doi: 10.1016/S0092-8674(00)81420-8. [DOI] [PubMed] [Google Scholar]
- 49.Hammes SR, Shapiro MJ, Coughlin SR. 1999. Shutoff and agonist-triggered internalization of protease-activated receptor 1 can be separated by mutation of putative phosphorylation sites in the cytoplasmic tail. Biochemistry 38:9308–9316. doi: 10.1021/bi9902236. [DOI] [PubMed] [Google Scholar]
- 50.Essalmani R, Hamelin J, Marcinkiewicz J, Chamberland A, Mbikay M, Chretien M, Seidah NG, Prat A. 2006. Deletion of the gene encoding proprotein convertase 5/6 causes early embryonic lethality in the mouse. Mol Cell Biol 26:354–361. doi: 10.1128/MCB.26.1.354-361.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dubois CM, Blanchette F, Laprise MH, Leduc R, Grondin F, Seidah NG. 2001. Evidence that furin is an authentic transforming growth factor-beta1-converting enzyme. Am J Pathol 158:305–316. doi: 10.1016/S0002-9440(10)63970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. 2002. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto K, Loskutoff DJ. 1998. Extrahepatic expression and regulation of protein C in the mouse. Am J Pathol 153:547–555. doi: 10.1016/S0002-9440(10)65597-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong W, Marcinkiewicz M, Vieau D, Chretien M, Seidah NG, Day R. 1995. Distinct mRNA expression of the highly homologous convertases PC5 and PACE4 in the rat brain and pituitary. J Neurosci 15:1778–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen DG, Wolff KC, Yin H, Caldwell JS, Kuhen KL. 2006. “UnPAKing” human immunodeficiency virus (HIV) replication: using small interfering RNA screening to identify novel cofactors and elucidate the role of group I PAKs in HIV infection. J Virol 80:130–137. doi: 10.1128/JVI.80.1.130-137.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutierrez-Rodriguez M, Herranz R. 2015. From multiple PAR1 receptor/protein interactions to their multiple therapeutic implications. Curr Top Med Chem 15:2080–2114. doi: 10.2174/1568026615666150519103911. [DOI] [PubMed] [Google Scholar]