

ABSTRACT
The emergence of Middle East respiratory syndrome-coronavirus (MERS-CoV) in the Middle East since 2012 has caused more than 900 human infections with ∼40% mortality to date. Animal models are needed for studying pathogenesis and for development of preventive and therapeutic agents against MERS-CoV infection. Nonhuman primates (rhesus macaques and marmosets) are expensive models of limited availability. Although a mouse lung infection model has been described using adenovirus vectors expressing human CD26/dipeptidyl peptidase 4 (DPP4), it is believed that a transgenic mouse model is needed for MERS-CoV research. We have developed this transgenic mouse model as indicated in this study. We show that transgenic mice globally expressing hCD26/DPP4 were fully permissive to MERS-CoV infection, resulting in relentless weight loss and death within days postinfection. High infectious virus titers were recovered primarily from the lungs and brains of mice at 2 and 4 days postinfection, respectively, whereas viral RNAs were also detected in the heart, spleen, and intestine, indicating a disseminating viral infection. Infected Tg+ mice developed a progressive pneumonia, characterized by extensive inflammatory infiltration. In contrast, an inconsistent mild perivascular cuffing was the only pathological change associated with the infected brains. Moreover, infected Tg+ mice were able to activate genes encoding for many antiviral and inflammatory mediators within the lungs and brains, coinciding with the high levels of viral replication. This new and unique transgenic mouse model will be useful for furthering knowledge of MERS pathogenesis and for the development of vaccine and treatments against MERS-CoV infection.
IMPORTANCE Small and economical animal models are required for the controlled and extensive studies needed for elucidating pathogenesis and development of vaccines and antivirals against MERS. Mice are the most desirable small-animal species for this purpose because of availability and the existence of a thorough knowledge base, particularly of genetics and immunology. The standard small animals, mice, hamsters, and ferrets, all lack the functional MERS-CoV receptor and are not susceptible to infection. So, initial studies were done with nonhuman primates, expensive models of limited availability. A mouse lung infection model was described where a mouse adenovirus was used to transfect lung cells for receptor expression. Nevertheless, all generally agree that a transgenic mouse model expressing the DPP4 receptor is needed for MERS-CoV research. We have developed this transgenic mouse model as indicated in this study. This new and unique transgenic mouse model will be useful for furthering MERS research.
INTRODUCTION
A severe acute respiratory syndrome caused by a new coronavirus (SARS-CoV) emerged in late 2002 and spread throughout Asia and to North America (http://www.who.int/csr/sars/country/table2004_04_21/en/). The infection caused ∼8,000 cases of SARS in humans with ∼10% mortality before the epidemic was brought under control by using infection control measures (1). Investigations concluded that the new virus (SARS-CoV) likely emerged from bats and that an intermediate animal host (civets) might have been involved in spread (2, 3). Ten years later (in 2012), another new CoV emerged in the Middle East as a cause of SARS-like disease in humans and was named Middle East respiratory syndrome (MERS)-CoV (4, 5). The outbreak of MERS-CoV is ongoing and, as of 5 November 2014, has caused 929 laboratory-confirmed cases with 372 deaths (∼ 40% mortality rate). The natural reservoir of MERS-CoV is thought again to be bats, with camels as a possible intermediate host (6, 7).
Although much information has been gained about MERS and MERS-CoV since emergence, significant deficiencies in basic and translational research exist, largely stemming from lack of suitable small-animal models for studies of pathogenesis and for development of vaccines and antivirals. Mice are the most desirable small animal for this purpose because of availability and the existence of a vast knowledge base, particularly of genetics and immunology. Unfortunately, standard small animals used in research such as mice, hamsters, and ferrets, all lack the MERS-CoV receptor, human CD26 (hCD26), or dipeptidyl peptidase-4 (DPP4) and are not susceptible to MERS-CoV infection (8–11). Although studies with nonhuman primates (NHPs) such as rhesus macaques and marmosets have demonstrated their susceptibility to various degrees of MERS-CoV infection, NHPs are expensive models with limited availability (12, 13). Although a mouse lung infection model was described wherein an adenovirus vector was used to transduce the viral receptor gene (14), it is generally agreed that a transgenic mouse model expressing the hCD26/DPP4 receptor is needed for research on MERS-CoV infection and disease. To derive such models, we used constitutive global or tissue-specific promoters, as well as an inducible, tissue-specific promoter. We describe here the first transgenic mouse lineage globally expressing hCD26/DPP4 receptor and show that these transgene-positive (Tg+), but not transgene-negative (Tg−), mice are highly susceptible to MERS-CoV infection and disease.
MATERIALS AND METHODS
Construction and characterization of a hCD26 expressing plasmid in vitro.
The transgene cassette expressing hCD26 (also known as dipeptidyl peptidase-4 [DPP4]) was constructed using pCAGGS.MCS, a eukaryotic expression vector, as we previously described in making a construct expressing the hACE-2 receptor of SARS-CoV (15). Briefly, cDNA of hCD26, the viral receptor of MERS-CoV, which was generated from the mRNA of human phytohemagglutinin-activated T cells (16) and provided to us by C. Morimoto, University of Tokyo, Tokyo, Japan, was cloned into pCAGGS.MCS under the control of the CAG promoter, which is a composite promoter consisting of the cytomegalovirus (CMV)/immediate-early enhancer and the chicken β-actin promoter, containing rabbit globulin splicing and polyadenylation sites. This CAG promoter was chosen because it is a strong synthetic promoter frequently used to derive high levels of gene expression in mammalian expression vectors. In addition, it has an advantage over the first-generation CMV promoter in that its activity does not decline over generations of transgenic mouse breeding, as do some other CMV-based promoters (17). To verify the transgene construct, designated pCAGGS-hCD26 (Fig. 1A), we transfected 17CL-1 mouse fibroblast cells and assessed the expression of hCD26 protein by Western blotting analyses using a goat polyclonal antibody against hCD26 known to have ∼5% cross-reactivity to mouse CD26 (R&D Systems). Established hCD26-expressing mouse 17CL-1 cells, along with cells transfected with empty vector, were tested for MERS-CoV susceptibility to verify the permissiveness of hCD26-expressing cells to infection by monitoring yields of progeny virus and the formation of cytopathic effect (CPE).
FIG 1.

Transfection with the hCD26 transgene construct, pCAGGS.hCD26, effectively converts nonsusceptible mouse fibroblastic 17CL-1 cells to become fully supportive of productive MERS-CoV infection. (A) Schematic diagram of hCD26 expressing vector cassette, designated pCAGGS.hCD26. The hCD26 gene was cloned in this vector by restriction digestion with EcoRI at the multiple cloning site (MCS) which is driving the expression in the presence of chicken β-actin promoter. Introns and polyadenylation signals were included in the expressing cassette to enhance the transcription and increase the mRNA stability of transgenes. These elements were cloned into the plasmid such that the transgene could be expressed in one piece from the plasmid backbone by digestion with restriction enzymes (38, 39). Confluent parental and 17CL-1 cells stably transfected with the hCD26 transgene construct were analyzed for the expression of hCD26 with a goat anti-hCD26 antibody using indirect immunofluorescent staining (B) and Western blot analysis (C). The expression of hCD26 antigen was detected exclusively in 17CL-1/hCD26 cells as the green fluorescent protein with an expected size of 110 kDa. (D to F) To assess the permissiveness of 17CL-1/hCD26 cells to MERS-CoV infection, confluent 17CL-1 and 17CL-1/hCD26 cells, grown in 12-well plates, were infected with either MERS-CoV/EMC-2012 or rMERS-CoV/RFP at an MOI of 1 and monitored for CPE for 2 days after infection (D and E), and yields of infectious progeny virus were titrated at 1 and 2 dpi (F; the dashed line indicates the detection limit). The data shown are representative of at least two independently conducted experiments. The infectious virus titer (log10 TCID50/ml) is expressed as the mean ± the standard error of triplicate samples. ***, P ≤ 0.001 (determined using the Student t test, comparing 17CL-1 and 17CL-1/hCD26 cells).
Generation, detection, and breeding of transgenic mice.
The transgene (∼5.6 kb) comprising CAGG enhancer/promoter, intron sequence, human CD26 cDNA, and rabbit beta globin poly(A) signal was released from pCAGGS.hCD26 using SalI and AvrII restriction enzymes and injected into B6C3F1/J × C57BL/6J or C57BL/6J zygotes. G0 founder mice were tested for transgene integration using quantitative PCR (qPCR) and/or Southern blot analysis. Briefly, tail genomic DNA isolated from tail biopsy specimens was subjected to qPCR using hCD26-specific primers (forward, 5′-CCAAAGACTGTACGGGTTCC-3′; reverse, 5′-TCAACATAGAAGCAGGAGCAG-3′) and fluorescence probe (5′-/56-FAM/AAGGCAGGAGCTGTGAATCCAACT/36-TAMSp/-3′) on a C1000 Touch thermocycler linked to a CFX96 real-time detection system (Bio-Rad). In some cases, Southern blot analysis was used to identify transgene-positive founder mice; for this, tail DNA was digested by BamHI, separated on agarose gels, and transferred to Hybond-XL (GE Healthcare Life Sciences). The blots were hybridized with 32P-labeled probes prepared by random primed labeling. A 0.4-kb SalI-NcoI fragment of the CMV enhancer and a 0.7-kb BglII fragment (DPP4 3′ untranslated region [3′UTR]), isolated from pCAGGS.hCD26, were used as probes. The Gsc2 5′ probe was used to normalize the amount of DNA on the blots (18). The transgenic mouse experiments were carried out in the barrier facility of the University of Texas Medical Branch transgenic mouse core facility. All animal work conformed to National Institutes of Health (NIH) and AAALAC regulations and guidelines.
Virus and cells.
The EMC-2012 strain of MERS-CoV, kindly provided by Heinz Feldmann (NIH, Hamilton, MT) and Ron A. Fouchier (Erasmus Medical Center, Rotterdam, Netherlands), was used throughout the present study. Vero E6 cells (American Type Culture Collection) were used to expand the virus stocks and titrate the yields of progeny viruses. Briefly, confluent Vero E6 cells were inoculated with MERS-CoV/EMC-2012 (passage 2) at a multiplicity of infection (MOI) of 0.001 to generate the passage 3 working stock of the virus. The cell-free supernatants derived from infected cultures were titrated using Vero E6 cell-based infectivity assays and expressed as 50% tissue culture infectious dose (TCID50)/ml. Aliquots of virus stock with a typical titer of ∼107 TCID50/ml were stored at −80°C. In addition, a strain of recombinant MERS-CoV expressing red fluorescent protein (rMERS-CoV/RFP), kindly provided by Ralph Baric, University of North Carolina at Chapel Hill (11), was similarly expanded and used in the indicated experiments to visualize the infection. All experiments involving infectious virus were conducted at the University of Texas Medical Branch (Galveston, TX) in approved biosafety level 3 (BSL-3) laboratories and animal facilities, with routine medical monitoring of staff.
Viral challenge and morbidity and mortality studies of infected mice.
All animal experiments were carried out in accordance with animal protocols approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch. Since the result of a pilot study indicated that hCD26 transgene-positive (Tg+) mice of different genetic background were equally susceptible to MERS-CoV infection (data not shown), Tg+ and their age-matched transgene-negative (Tg−) littermates derived from the line 52 (see description below) founder mouse were backcrossed one or two times onto either a C57BL/6 or B6C3F1/J background. Anesthetized Tg+ and Tg− littermates at ages 5 to 7 weeks were inoculated via the intranasal (i.n.) route with 106 TCID50 of MERS-CoV in a total volume of 80 μl. Animals were weighed and monitored daily for clinical signs of disease and abnormalities, including appearance, stereotypical behavior/abnormal movements, decreased responsiveness or activity, and weight loss. The clinical scoring system used was as follows: 0, no apparent illness; 1, mildly sick; 2, ruffled fur or hunching; 3, ruffled fur and hunching, with or without additional signs; 4, moribund; and 5, found dead. Some infected mice were sacrificed at indicated times postinfection (p.i.) to obtain tissue specimens for assessing the distribution of virus and associated histopathology using standard protocols, such as a Vero E6 cell-based infectivity assay, quantitative reverse transcription-PCR (qRT-PCR) assay, and immunohistochemical (IHC) staining.
Virus isolation.
Pieces of collected tissue specimens (lungs, brain, heart, liver, kidney, spleen, and intestine) were weighed and homogenized in phosphate-buffered saline (PBS)–10% fetal calf serum solution using a TissueLyser (Qiagen, Retsch, Haan, Germany) to yield 10% tissue–PBS suspensions. These suspensions were clarified by centrifugation and tested in a Vero E6 cell-based assay for titrating quantity of infectious virus. The virus titers of individual samples were expressed as TCID50 per g of tissue.
RNA extraction and real-time quantitative qRT-PCR.
Pieces of tissues were transferred to individual vials containing RNAlater solution (Qiagen) and subsequently homogenized as described previously and subjected to total RNA isolation, using TRIzol Reagent (Life Technologies), for assessing MERS-CoV-specific genome targeting the virus-specific upstream E gene (upE) and mouse GAPDH gene (mGAPDH), as previously described (19). For the detection of hCD26 and viral gene expression in different tissues, 0.5 μg of RNA was used in a one-step real-time RT-PCR with a gene-specific primer/probe mix for the hCD26 gene and upE gene of MERS-CoV, using the Superscript III One-step RT-PCR kit (Invitrogen) according to the manufacturer's instructions. The primers and probes used for upE gene of MERS-CoV were as follows: forward, 5′-GCC TCT ACA CGG GAC CCA TA-3′; reverse, 5′-GCA ACG CGC GAT TCA GTT-3′; and fluorescence probe, 5′-/56-FAM/CTC TTC ACA TAA TCG CCC CGA GCT CG/36-TAMSp/-3′. The relative amount of targeted mRNA was obtained by normalizing with endogenous control gene (mGAPDH) and calculated in terms of the fold difference by the standard threshold cycle (ΔΔCT) method (20).
Transcriptional profiling of cytokine and chemokine responses by qRT-PCR assays.
RNA extracted from either lung or brain tissue was subjected to qRT-PCR analyses for quantifying transcriptional expression of selected genes encoding key cytokines and chemokines. Briefly, 1 μg of RNA from an individual tissue obtained from infected Tg+ or Tg− animals was converted to cDNA in a 20-μl reaction volume containing Superscript II (Invitrogen, Carlsbad, CA), oligo(dT) (500 ng), 20 U of RNase OUT, and 10 mM deoxynucleoside triphosphate mix in a 20-μl reaction volume. The reaction conditions were strictly according to the manufacturer's instructions. Transcription profiles of each cytokine/chemokine in the brain and lung were determined by real-time qPCR using iQ SYBR green PCR Supermix (Bio-Rad). Real-time reactions were set up in duplicate for each cytokine or chemokine using endogenous mouse GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene as internal controls for both brain and lung tissues using specific primer sets in Table 1. An identical amplification reaction condition consisting of (i) polymerase activation and DNA denaturation at 95°C for 3 min, (ii) 40 cycles each of the denaturation step at 95°C for 10 s, and (iii) an annealing/extension step at 60°C for 30 s was used for each gene analyzed. The reaction volume was kept at 10 μl by including 1 μl (10 ng) of cDNA from these reactions. The relative abundance of transcripts of each gene was calculated according to the comparative ΔΔCT method (20).
TABLE 1.
Primers of selected antiviral and inflammatory genes
| Primer | Gene | Primer | Region | Amplicon size (bp) | |
|---|---|---|---|---|---|
| Orientation | Sequence (5′-3′) | ||||
| 1 | IL-1β | F | TGG ACC TTC CAG GAT GAG GAC A | 897–1044 | 148 |
| R | GTT CAT CTC GGA GCC TGT AGT G | ||||
| 2 | IL-2 | F | GGA CTT TCT GAG GAG ATG GAT AG | 498–581 | 84 |
| R | TGT TGT AAG CAG GAG GTA CAT AG | ||||
| 3 | IL-6 | F | TAC CAC TTC ACA AGT CGG AGG C | 169–284 | 116 |
| R | CTG CAA GTG CAT CAT CGT TGT TC | ||||
| 4 | IL-12p40 | F | TTG AAC TGG CGT TGG AAG CAC G | 720–851 | 132 |
| R | CCA CCT GTG AGT TCT TCA AAG GC | ||||
| 5 | TNF-α | F | TTG TCT ACT CCC AGG TTC TCT | 526–632 | 107 |
| R | GAG GTT GAC TTT CTC CTG GTA TG | ||||
| 6 | G-CSF | F | GCA GGC TCT ATC GGG TAT TTC | 1895–2006 | 112 |
| R | CAC CCC TAG GTT TTC CAT CTG | ||||
| 7 | MCP-1 | F | GTC CCT GTC ATG CTT CTG G | 92–235 | 144 |
| R | GCT CTC CAG CCT ACT CAT TG | ||||
| 8 | IP-10 | F | ATC ATC CCT GCG AGC CTA TCC T | 223–356 | 134 |
| R | GAC CTT TTT TGG CTA AAC GCT TTC | ||||
| 9 | CXCL-1(KC) | F | TCC AGA GCT TGA AGG TGT TGC C | 206–315 | 110 |
| R | AAC CAA GGG AGC TTC AGG GTC A | ||||
| 10 | MIP-1α | F | ACC ATG ACA CTC TGC AAC C | 141–243 | 103 |
| R | CGA TGA ATT GGC GTG GAA TC | ||||
| 11 | RANTES | F | CCT GCT GCT TTG CCT ACC TCT C | 152–276 | 125 |
| R | ACA CAC TTG GCG GTT CCT TCG A | ||||
| 12 | IFN-β | F | GCG GAC TTC AAG ATC CCT ATG | 183–322 | 140 |
| R | ACA ATA GTC TCA TTC CAC CCA G | ||||
| 13 | IFN-γ | F | AAA TCC TGC AGA GCC AGA TTA T | 299–426 | 128 |
| R | GCT GTT GCT GAA GAA GGT AGT A | ||||
| 14 | MX-1 | F | TGG ACA TTG CTA CCA CAG AGG C | 773–887 | 115 |
| R | TTG CCT TCA GCA CCT CTG TCC A | ||||
Histology and IHC testing.
Tissues obtained from necropsy samples were fixed in 10% buffered formalin for 72 h, transferred to 70% ethanol, and later paraffin embedded. Histopathologic evaluation was performed on deparaffinized sections stained by routine hematoxylin-eosin (H&E) staining. IHC testing for MERS-CoV was performed using a previously described colorimetric indirect immunoalkaline phosphatase method (15) with a rabbit anti-MERS-CoV polyclonal antibody, a gift from Heinz Feldmann. The goat anti-hCD26 antibody (R&D Systems, catalog no. AF1180) was used to assess the distribution of hCD26 expression in transgenic mice by IHC analysis. Normal mouse and goat sera were used as negative antibody controls. Primary antibodies were detected with either biotinylated swine anti-rabbit immunoglobulin (Dako, catalog no. E0353) or rabbit anti-goat immunoglobulin (KPL, catalog no. 16-13-06) as secondary antibodies. Visualization was then achieved by incubation with streptavidin-alkaline phosphatase and naphthol-fast red substrate (Dako) and with counterstaining with Mayer's hematoxylin (Fisher Scientific).
RESULTS
Characterization of the hCD26 transgene construct in tissue culture.
To validate the potential of the hCD26 transgene cassette (Fig. 1A) in expressing the transgene, mouse fibroblastic 17 CL-1 cells were cotransfected with the hCD26 expression plasmid, pCAGGS-CD26, and a plasmid encoding puromycin resistance. After selection with puromycin (2 μg/ml), the transfectants were assessed for level of transgene expression at the protein level by both immunofluorescence (IF) staining and Western blotting analyses. As shown in Fig. 1B and C, an intense green fluorescence staining and a distinct band of ∼110 kDa in Western blots, the estimated size of hCD26, were seen almost exclusively in 17CL-1 cells transfected with the hCD26 expressing plasmid, indicating the effectiveness of the transgene construct in promoting cellular hCD26 expression. The much weaker band seen in wild-type 17CL-1 cells likely represented mouse CD26 as the anti-hCD26 antibody (R&D Systems) used for Western blotting was shown to having ∼5% cross-reactivity to mouse CD26. To determine whether the expression of hCD26 viral receptor would convert nonpermissive 17CL-1 cells to cells that support MERS-CoV infection, confluent cultures of 17CL-1/hCD26 cells and parental 17CL-1 cells were infected with either MERS-CoV or a recombinant MERS-CoV expressing red fluorescent protein (RFP) at an MOI of 1, followed by monitoring for morphological changes of infected cells and for the intensity of infection over time. In contrast to the infection-resistant parental 17CL-1 cells, 17CL-1/hCD26 cells were fully susceptible to MERS-CoV infection, resulting in the development of CPE (Fig. 1D), readily detectable expression of RFP (Fig. 1E), and high yields of infectious progeny virus as early as 1 day postinfection (dpi) (Fig. 1F). Based on these results, we conclude that induction of hCD26 expression using this transgene construct effectively converts nonpermissive 17CL-1 cells into permissive cells, capable of fully support MERS-CoV infection.
Generation and characterization of hCD26 transgenic mice.
Transgenic mice expressing hCD26 were generated by microinjecting the expression cassette, excised from pCAGGS-CD26 by AvrII/SalI digestion, into pronuclei of zygotes from either the C57BL/6J or C57BL/6J × B6C3F1/J background, as described in Materials and Methods, which led to 81 live births. Based on the qPCR and/or Southern blot analyses of tail DNA, we identified five B6/C3H hybrid and two B6 founders that were then crossed with C57BL/6J and/or B6C3F1/J to propagate the lines. Among seven Tg+ founder lineages, lines 52, 62, and 72 were selected, based on their ability to transmit hCD26 transgene to offspring, to further characterize hCD26 expression and expand the transgenic colonies. Southern blot analyses revealed that the intensity of hCD26 transgene was greatest in line 62, followed by line 72 and line 52, in that order (Fig. 2A). Line 62 suffered from both extremely poor transgene transmissibility and neonatal deaths of Tg+ pups, whereas the majority of Tg+ pups of Line 72 died prematurely at ∼3 weeks of age (Table 2). To evaluate the expression of hCD26 in lines 72 and 52, total RNA was extracted from tissues of Tg+ G1 pups and subjected to qRT-PCR analysis using the same hCD26-specific primer-probe set used for tail genotyping. Although both lines expressed hCD26 in all tissues analyzed, the levels of hCD26 expression appeared to be higher in line 72 than in line 52, with the heart and spleen as the only exceptions (Fig. 2B).
FIG 2.

Expression of hCD26 in transgene-positive (Tg+) mice. (A) Genomic DNA extracted from tail biopsy specimens of hCD26 transgenic founder mice of lines 52, 62, and 72 were digested by BamHI and subjected to Southern blot analyses to determine the relative intensities of the integrated hCD26 transgene, as described in Materials and Methods. The relative intensities of hCD26 among these three Tg+ lineages are presented as ratios of the signals probed by a SalI-NcoI fragment (CMV enhancer) and a 0.7-kb BglII fragment (DPP4 3′UTR, isolated from pCAGGS.hCD26, and Gsc2 5′) and used to normalize the amount of DNA. (B) Tg+ mice derived from lines 52 and 72 were euthanized, and their various tissue specimens were harvested to extract total RNA to assess the relative tissue abundances of hCD26 mRNA expression by qRT-PCR, as described in Materials and Methods. (C) hCD26 Tg+ mice of line 52 were euthanized, and the homogenates and paraffin-embedded sections of various tissues were prepared to assess the expression of hCD26 protein by Western blotting with the anti-hCD26 antibody described in Fig. 1. A varying intensity of hCD26 protein was detected in all of the tissues analyzed. (D) Paraffin-embedded tissues derived from Tg+ and Tg− mice were subjected to IHC staining with an anti-hCD26 antibody to localize hCD26 expression. In Tg+ mice, the expression of hCD26 in the lungs was readily detectable in type I and type II alveolar pneumocytes (arrow), endothelial cells (arrow), and neurons (arrowhead) in the brain, muscle cells (arrow) in the heart, endothelial cells (arrow) and the surface of hepatocytes (arrowhead) in the liver, endothelial cells (arrow) in the kidneys, and epithelial lining (arrowhead) and the underneath muscularis (arrow) in the intestines. No hCD26 expression was evident in the tissues of Tg− mice.
TABLE 2.
Founders of hCD26 transgenic lineagesa
| Founder (lineage) | Sex | Background | Transgene confirmation method | Germ line transmission | Notes |
|---|---|---|---|---|---|
| 7 | F | B6/C3 mix | SB/qPCR | No | The mouse was fertile but produced no transgene-positive pups. |
| 52 | F | B6/C3 mix | qPCR | Yes | The mouse was viable and fertile. |
| 54 | M | B6/C3 mix | qPCR | ND | The mouse died shortly after weaning. |
| 62 | M | B6/C3 mix | SB/qPCR | Yes | The mouse was fertile but produced small litters. A few transgene-positive pups were found, but they died within a week after birth. |
| 72 | F | B6/C3 mix | SB/qPCR | Yes | The mouse was fertile. The majority of transgene-positive pups died after weaning before achieving sexual maturity. |
| 76 | ND | B6 | SB/qPCR | ND | The mouse died before weaning. |
| 78 | F | B6 | SB/qPCR | Yes | The mouse was fertile but did not take care of the pups. None of the pups survived beyond neonatal periods. |
ND, not determined; SB, Southern blotting; B6/C3 mix, crossed with B6C3F1/J and C57BL/6J.
In contrast to the extreme difficulty that we encountered to propagate lines 62 and 72, the line 52 founder mouse was capable of generating many first-, second-, and third-generation Tg+ pups that survived to maturity, permitting characterizing of hCD26 expression. To investigate the tissue distribution of hCD26 protein expression, cellular lysates and paraffin-embedded sections of various tissues were analyzed by Western blotting and IHC staining using a polyclonal antibody known to recognize hCD26 with <5% cross-reactivity to mouse CD26 (R&D Systems, catalog no. AF1180). Among six tissues analyzed (i.e., heart, lung, spleen, intestine, liver, and kidneys) by Western blotting, the expression of hCD26 was higher in both lungs and kidneys than in other tissues (Fig. 2C). The standard IHC assays also detected the expression of hCD26 antigen in all tissues analyzed, including the lung, brain, heart, liver, kidney, and intestine (Fig. 2D). We noted in Tg+ mice that hCD26 was primarily detected in both types of alveolar pneumocytes in lung and neuronal cells and endothelial cells in brain (Fig. 2Da and b). Prominent epithelial and/or endothelial hCD26 expression was detected in the liver, kidneys, and the gastrointestinal (GI) tract (Fig. 2Dd to f). Hepatic expression of hCD26 extended to the surface of hepatocytes. The expression of hCD26 was focalized within the muscularis layer of the GI tract and cardiomyocytes of the heart. Importantly, this positive staining is specific to hCD26 since no staining could be detected in tissues from Tg− mice. Taken together, these results indicate that line 52 is a stable transgenic lineage globally expressing the hCD26 receptor for MERS-CoV.
Transgenic mice expressing hCD26 are permissive to MERS-CoV infection that results in disease and mortality.
With the characterized hCD26 transgenic lineage, we explored whether line 52 hCD26 Tg+ mice would be permissive to MERS-CoV infection. For a pilot study, we infected i.n. two Tg+ mice of different genetic backgrounds obtained from backcrossing to either B6 or B6C3F1/J mice, along with two Tg− age-matched littermates, with 106 TCID50 of MERS-CoV in 80 μl. Mice were sacrificed 2 days later after infection to assess the yield of infectious virus in the lungs using Vero E6 cell-based infectivity assays. We found that Tg+ mice of different genetic background, but not those of Tg− littermates, were equally susceptible to infection, as evidenced by high yields of infectious virus (data not shown).
Encouraged by the results of this pilot study, we subsequently inoculated additional age-matched line 52 Tg+ and Tg− mice; nine animals in each Tg group were given 106 TCID50 i.n. in 80 μl for initial assessment of the kinetics of infection and disease. The tissue distribution of infection and disease was assessed by sacrificing two mice in each group at 2 and 6 dpi. Mice were monitored daily for signs of clinical illness, weight loss, and mortality. We noted that challenged Tg+, but not Tg−, mice developed an acute wasting syndrome, as evidenced by progressive weight loss starting at 2 dpi, reaching 30% at 5 dpi, and leading subsequently to 40 and 100% mortality at 5 and 6 dpi, respectively (Fig. 3A and B). Other clinical manifestations of infected Tg+ mice included ruffled fur, lethargy, inactivity, and rapid and shallow breathing. Despite their immobility, we did not observe any signs of neurological disorder, such as seizure or paralysis, in the Tg+ mice that were severely affected by MERS-CoV infection. The hCD26 Tg− mice continued to thrive throughout the course of infection without showing any weight loss or signs of clinical illness.
FIG 3.

Transgenic mice expressing hCD26 are permissive to MERS-CoV infection, leading to morbidity and mortality. Both hCD26 Tg+ and Tg− mice, nine animals each, were challenged i.n. with 106 TCID50 of MERS-CoV in 80 μl. Infected mice were monitored daily for their weight gain or loss (A), other signs of clinical illness, and mortality (B). Two animals were euthanized at 2 and 4 dpi to determine the viral loads in various tissues harvested by quantifying infectious virus isolated (C) and viral RNA targeting upstream E gene by qRT-PCR (D). In addition, paraffin-embedded lung (E and F) and brain (G and H) tussues were stained for MERS-CoV antigen with a specific anti-MERS-CoV antibody raised in rabbits, as described in Materials and Methods. (A) Persistent weight loss (up to 30%) in infected Tg+, but not Tg−, mice. (B) Cumulative survival rate of infected mice. Tg+ mice succumbed to infection with 100% mortality at 6 dpi. (C) Infectious virus titers, expressed as log10 TCID50 per g of tissue, in the lungs and brain. (D) Copy numbers of viral E gene in the indicated tissues of Tg+ mice, relative to those derived from Tg− littermates. (E and F) Viral antigen (red) was readily detected in alveolar pneumocytes of Tg+ (E), but not Tg− (F), mice at 2 dpi. (G and H) Viral antigen (red) was also detected in neurons (green arrow), microglia (arrowhead), astrocytes (black arrow), and astrocyte processes (star) in the brains of Tg+ (G), but not Tg− (H), mice at 4 dpi.
Line 52 transgenic mice expressing hCD26 developed disseminated infection.
To determine the kinetics and tissue distribution, tissue specimens of Tg+ and Tg− mice collected on 2 and 4 dpi were tested for infectious virus in the Vero E6 cell-based infectivity assay. It was clear that lung and brain appeared to be the prime sites of intense viral infection (Fig. 3C and D). We detected titers as high as ∼107 TICD50/g of lung at 2 dpi that dropped at 4 dpi to ∼104 TCID50/g, a net loss of 3 logs. In contrast to the acute and robust pulmonary infection, we were unable to isolate infectious virus from the brain until 4 dpi when an average of 7 × 104 TCID50/g was detected. These results indicate that the kinetics of MERS-CoV infection in the lung and brain of Tg+ mice were substantially different. Despite the high yields of infectious virus detected in the lung and brain, we were unable to isolate any infectious virus from other tissues, including the liver, heart, spleen, kidneys, and intestines, at either 2 or 4 dpi (data not shown). To verify the presence or absence of virus in tissues with no infectious virus, we tested for virus in all tissues collected at both 2 and 4 dpi for the presence of viral RNA by qRT-PCR targeting the upstream E gene of MERS-CoV. Consistent with the high yields of infectious virus, a 6-log-greater level of specific viral RNA was detected in the lungs and brains of infected Tg+ mice than in Tg− mice (Fig. 3D). Importantly, MERS-CoV-specific viral RNA was also readily detected at either 2 or 4 dpi in the heart, spleen, and intestine, although we were not able to isolate any infectious virus, suggesting that a disseminated MERS-CoV infection had occurred in the hCD26 Tg+ mice. Interestingly, however, we were unable to detect any viral RNA in the liver and kidneys, despite the expression of hCD26 in these organs (Fig. 2B, C, and Dd and e).
Because high titers of infectious virus could be readily recovered from lungs and brains of challenged Tg+ mice, we performed IHC staining with paraffin-embedded tissues and a specific antibody for detecting viral antigen, as described in Materials and Methods, to confirm the cellular tropism of MERS-CoV infection for each tissue. As shown in Fig. 3E and G, lung alveolar pneumocytes, both type I and type II, and brain microglia, astrocytes, and neuronal cells expressed abundant viral antigen in Tg+ mice at 2 and 4 dpi, a finding consistent with the high titers of virus. These patterns of viral antigen expression were compatible with the cellular distribution of the hCD26 receptor for MERS-CoV (Fig. 2D), even though a two-color IHC staining was not performed. These findings indicate that transgenic mice expressing hCD26/DPP4 are highly susceptible to a MERS-CoV infection that results in significant morbidity and mortality.
Histopathology in MERS-CoV-infected hCD26 Tg+ mice.
Pathological changes in virally challenged Tg+ and Tg− mice were assessed at 2 and 4 dpi. Consistent with the intense viral infection, gross lesions, consisting of discoloration (red to dark red) and multifocal consolidation, were observed in the lungs of Tg+ but not in Tg− mice at 2 dpi, and changes had progressed at 4 dpi (Fig. 4A). Histological examination of the lung at 2 dpi showed a moderate bronchointerstitial pneumonitis and multifocal perivascular infiltrates with some changes extending into terminal bronchioles and adjacent pulmonary parenchyma. At 4 dpi, more intense cellular infiltrates, including pulmonary macrophages and lymphocytes, were seen within alveolar spaces (Fig. 4B). Both type I and type II alveolar epithelial cells appeared to be targets for MERS-CoV infection in hCD26 Tg+ mice (Fig. 2D). In contrast to the infected lungs, there was no necrosis or significant inflammatory reaction, other than mild perivascular cuffing identified in a single animal, observed in the brain, even at 4 dpi when the prominent viral infection was detected (Fig. 4C). Similarly, no pathological findings could be identified in other tissues collected from virus-challenged Tg+ or Tg− mice (data not shown).
FIG 4.
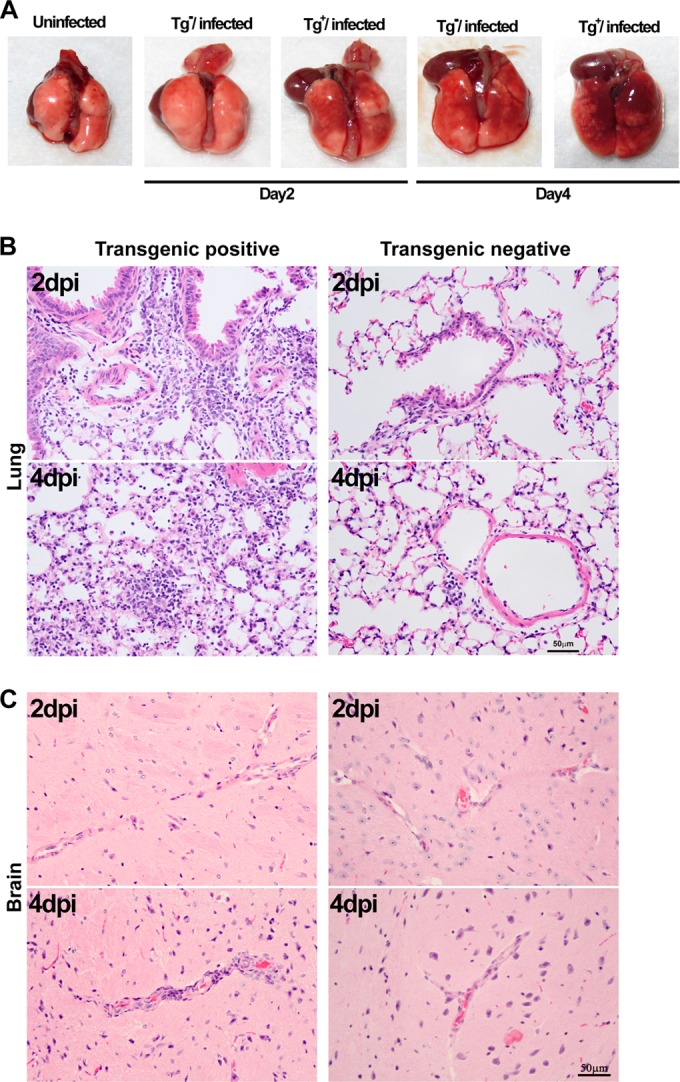
Histopathological changes in the lungs and brains of hCD26 Tg+ mice challenged with MERS-CoV. Tg+ and Tg− mice were euthanized on day 2 and 4 after challenge with MERS-CoV for assessing the pathology of the lungs and brain. (A) More extensive gross lesions of the lungs were detected in Tg+ mice than in Tg− mice, starting at day 2 after challenge. (B) Paraffin-embedded sections of lung and brain specimens were stained with H&E. Compared to a few foci of perivascular infiltration in the lungs of Tg− mice, a more intense and widespread bronchiolitis and alveolitis was observed in the lungs of Tg+ mice, starting at day 2 postchallenge. The major cell types of the inflammatory infiltrates were lymphocytes and monocytes, with a few scattered neutrophils.
MERS-CoV infection induces inflammatory responses in the lung and brain of Tg+ mice.
As noted for SARS-CoV infection, significant cytokine and chemokine responses in the lungs of patients severely affected by MERS-CoV infection have been implicated as important mechanisms of MERS pathogenesis (13, 14, 21). The rapid onset of morbidity and mortality of hCD26 Tg+ mice in response to respiratory MERS-CoV challenge prompted us to explore the host cytokine/chemokine responses in the lung and brain, the two most affected tissues. Specifically, we determined the transcriptional activation of genes encoding classic antiviral cytokines (IFN-β, IFN-γ, and MX-1) and proinflammatory cytokines (IL-2, IL-6, IL-12p40, IL-1β, and TNF-α), as well as chemokines (G-CSF, MCP-1, IP-10, CXCL-1, MIP-1α, and RANTES), using qRT-PCR. These 14 genes were selected on the basis of their elevated expression in the lung and brain, as demonstrated by us in hACE2 transgenic mice in response to respiratory challenge with SARS-CoV (15, 22). As shown in Fig. 5, compared to the negligible gene activation of infected Tg− mice, elevated mRNA levels of the majority of genes analyzed, including IL-1β, IL-6, TNF-α, G-CSF, MCP-1, IP-10, CXCL-1 (KC), MIP-1α, RANTES, and MX-1, were readily detected in both the lungs and brains of infected Tg+ mice in at least one time point evaluated (either 2 or 4 dpi) with few exceptions. Elevated mRNA expressions of both IFN-β and IFN-γ were observed exclusively within the lungs at 2 dpi, whereas activated IL-12p40-encoded gene could only be detected in the brain at 4 dpi. The IL-2-encoding gene was the only gene whose expression was not induced by MERS-CoV infection in Tg+ mice. We also noted that the magnitudes of gene activation in infected Tg+ mice were generally higher at 2 dpi in the lung and 4 dpi in brain, a pattern of response consistent with MERS-CoV replication within individual organs analyzed. Taken together, these results indicate not only that hCD26 Tg+ mice can fully support MERS-CoV infection, resulting in morbidity and mortality, but that they also respond to the infection by rapidly eliciting acute inflammatory responses.
FIG 5.

MERS-CoV infection induces profound acute innate inflammatory responses within the lungs and brain of hCD26 Tg+ mice. Total RNAs were extracted from the lungs and brains of Tg+ and Tg− mice at days 2 and 4 after infection with MERS-CoV and processed for profiling innate inflammatory responses by qRT-PCR, targeting a panel of 14 genes encoding antiviral and proinflammatory cytokines and chemokines. The relative abundance of mRNA transcripts of each gene was calculated according to the comparative ΔΔCT method. The results are presented as the fold changes in gene expression of infected Tg+ mice compared to that of infected Tg− mice. The data displayed represent the means ± the standard errors of two animals, with duplicate samples of each.
DISCUSSION
There is a need for small-animal models of the recently emerged MERS-CoV infection and disease so as to conduct studies of pathogenesis and immunity and for the development of vaccines and antivirals. The failure of mice, hamsters, and ferrets, the most commonly used animals for research, to support MERS-CoV infection (23) has impaired progress toward understanding this infection and disease and the development of vaccines and antivirals. This circumstance for MERS contrasts with that for SAR-CoV infection and disease, the earlier emerged severe acute respiratory disease wherein mice, hamsters, and other small animals were susceptible to infection and disease (24). Also, with passage in mice, a SARS-CoV virus strain was developed that caused severe respiratory disease and death (25). The virus receptor for SARS-CoV (hACE-2) is present in small animals and permitted a considerable research effort on SARS-CoV infection and disease. The virus receptor for MERS-CoV, i.e., CD26/DPP4, is not present on tissues of mice, hamsters, and ferrets, and these animals are not susceptible to infection, a circumstance considerably impairing research. Two NHP species, rhesus macaques and marmosets, and Ad5-hDPP4-transduced mice have been reported as models for MERS-CoV infection and disease (13, 14). In contrast to the transient and self-limited viral infection and mild-to-moderate respiratory disease that developed in rhesus macaques and Ad5-hDPP4-transduced mice, marmosets developed a more intense infection, predominantly in the respiratory tract, that caused progressive severe pneumonia and death in some animals. Currently, the marmoset appears to be the best model for MERS-CoV infection and disease.
With considerable effort, we succeeded in establishing a transgenic mouse model for MERS-CoV infection and disease. In this report, we present the characterization of this transgenic lineage with regard to hCD26/DPP4 transgene expression and the outcome of MERS-CoV challenge of transgenic mice. In contrast to the variable duration and level of hDPP4 expression, as well as the limited and transient disease developed by infected Ad5-hDPP4-transduced mice, we found that not only could these transgenic mice, globally expressing hCD26/DPP4, support a robust MERS-CoV infection, especially in lungs and brain, but they also developed a severe respiratory and generalized illness that led to death within days after infection. With several defined endpoints consistently seen in MERS-CoV-challenged hCD26/DPP4 transgenic mice, including death, relentless weight loss, and acute and profound pulmonary viral infection with lung pathological changes, we believe that this newly established transgenic mouse model will aide in the effort to better understand the pathogenesis of MERS-CoV infection and disease, as well as provide a more economical platform for preclinical evaluations of antivirals and vaccines for MERS.
Transgenic mice expressing hCD26/DPP4 were established using the pCAGGS.hCD26 plasmid that was first shown to convert nonpermissive mouse 17CL-1 cells to fully susceptible to MERS-CoV infection (Fig. 1). Among four identified fertile founders of transgenic lineages capable of transmitting the transgene to offspring (Table 1), three founders (lines 62, 72, and 78) suffered from either giving birth to very small litters or neonatal or the early death of pups. However, the line 52 founder was fertile and gave birth to litters with an average of 8 to 12 healthy pups, thereby allowing the generation of many G1, G2, and even G3 Tg+ offspring. Such a low rate in successfully establishing hCD26/DPP4 transgenic lineages was unexpected, as we had successfully used the same pCAGGS mammalian expression plasmid to establish at least five transgenic lineages globally expressing different levels of hACE-2 receptor for SARS-CoV (15), suggesting that the expression of hCD26/DPP4 may have negatively affected the well-being of mice.
It is well established that CD26/DPP4, a type II transmembrane serine peptidase, is unambiguously expressed at various intensities in many human tissues and can regulate diverse biological functions in health and disease through the cleavage of X-proline dipeptides of many growth factors, chemokines, neuropeptides, vasoactive peptides, and other biologically active polypeptides (26, 27). In addition, an ill-regulated enzymatic function of CD26/DPP4 could lead to the inactivation of incretin hormones, such as glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), resulting in decreasing glucose-dependent secretion of insulin, the hallmark of diabetes (28). Moreover, an increased CD26/DPP4 expression has been shown to negatively affect the growth of endothelial cells, especially within the microvasculature network (29). Therefore, DPP4 inhibitors have been used successfully as antidiabetic drugs, reversing the detrimental effect imposed by inactivated incretin hormones through increasing insulin secretion and suppressing glucagon secretion. Moreover, the intensity of CD26/DPP4 expression within the microvasculature network appears to negatively correlate with the growth capacity of endothelial cells, since the inhibition of CD26/DPP4 expression, afforded by TNF-α, or IL-1β treatment or other pharmacological manipulations, appeared to promote endothelial growth both in vitro and in vivo (29), providing the therapeutic basis for using DPP4 inhibitors for treating vascular complications associated with diabetes. Based on these reports of DPP4 function, we explored whether uncontrolled sugar metabolism could be responsible for neonatal or premature deaths of Tg+ pups of lines 62 and 72. Preliminary data obtained from a pilot study of line 62 founder and a few G1 or G2 pups (∼3 to 4 weeks of age) derived from lines 72 and 52 indicated that there were 2- to 3-fold increases in blood sugar levels in the Tg+ mice of each lineage compared to their Tg− littermates (data not shown). Coincidently, pathological examination also revealed the existence of irregular vasculature in the kidneys, lungs, and liver of Tg+ mice of Line 72 but not line 52 (data not shown). These results led us to speculate that hCD26 is likely catalytically active in Tg+ mice, having a negative impact, to a varying extent, on the well-being of mice in a lineage-dependent manner. However, whether such an “add-on” hDPP4 activity in Tg+ mice could be responsible for elevated blood sugar levels and abnormal vasculatures and contribute to breeding difficulties and prenatal or postnatal deaths awaits additional studies. Similarly, whether altered glucose metabolism influences the course of infection and disease in line 52 Tg+ mice awaits further study.
Studies of the kinetics and tissue distribution of MERS-CoV replication in virally challenged (i.n.) Tg+ mice derived from the only transgenic lineage established (line 52) clearly indicated that a robust viral infection took place primarily in the lung and brain, making them the prime tissues for MERS-CoV infection in these animals. The highest load of infectious virus in the lung was detected at 2 dpi and was followed by a decrease of 3 logs at 4 dpi, whereas infectious virus in the brain could not be detected until 4 dpi, suggesting that the respiratory tract was the initial site of MERS-CoV infection, which then spread to the brain (Fig. 3C). Studies of IHC for antigen expression showed that MERS-CoV primarily replicated in type I and II pneumocytes in the lung alveolae, whereas MERS-CoV infection in the brain could be detected in many cell types, including microglial, astrocytes, and neurons (Fig. 3E and G). Although infectious virus was not recovered from the other tissues collected, viral RNA was readily detected by qRT-PCR in the heart, spleen, and intestines, suggesting a disseminated infection had occurred. However, despite confirmed hCD26/DFPP4 expression, no viral RNA was detected in the liver and kidneys (Fig. 3D and 2D). The occurrence of acute renal failure has been reported to occur in some MERS patients (30) and viral loads in the kidneys of experimentally infected NHPs was evaluated, aiming at linking the acute kidney disease directly to MERS-CoV infection. Despite the widespread viral infection that occurred in experimentally infected rhesus macaques and marmosets, detection of viral RNA from kidneys could only be detected in marmosets; however, neither infectious virus nor histopathology was detected in the kidneys of infected marmosets (12, 13). As in our studies, respiratory infection with MERS-CoV in marmosets appeared to lead to virus in the central nervous system, as evidenced by the detection of viral RNA in the frontal lobe, cerebellum, and brain stem (13). Attempts to isolate virus from the brain were not made in infected marmosets by these investigators.
Histopathological examination of tissues collected from challenged Tg+ mice identified lungs as the tissue with the most pathological changes, a finding consistent with its supporting an acute and robust MERS-CoV infection. In addition to readily recognizable gross changes (Fig. 4A), microscopic lesions of infected lungs occurred as early as 2 dpi and progressed to an intense bronchointerstitial pneumonia at 4 dpi with prominent inflammatory infiltrates of lymphocytes, macrophages, and neutrophils (Fig. 4B). Although mild thickening of alveolar walls was present at 4 dpi, edema and/or fibrin deposition was not detected in pulmonary interstitium. Other than a mild perivascular cuffing in a single mouse, pathological change was not detected in the brain, even at 4 dpi when a robust viral replication was detected (Fig. 4C and Fig. 3). Similarly, no pathological changes were identified in the other tissues examined, i.e., the heart, liver, spleen, kidneys, and intestines (data not shown).
Although the exact mechanisms of contribution to the pathogenesis of MERS-CoV are unknown, an ill-regulated host innate inflammatory response has been implicated as responsible, at least in part, for the acute lung injury (13, 31). Based on the sites of intense viral replication identified in this transgenic mouse model of MERS-CoV infection and disease, we explored these host responses in the lungs and brain. As seen in experimentally challenged marmosets, robust innate antiviral and inflammatory responses were swiftly generated in the lungs and brains of Tg+ mice (Fig. 5). However, in contrast to downregulated expressions of IFN-γ and its receptor and undetectable IFN-β expression in marmoset lungs (13), elevated transcripts of both IFN-β and the IFN-stimulated gene MX-1 were detected in mouse lungs at 2 dpi. These expressions, along with those of other genes tested, returned to near the basal level of expression at 4 dpi. Among the 13 genes whose expressions were elevated in the infected lungs of Tg+ mice (IL-1β, IL-6, IL-12p40, TNF-α, G-CSF, MCP-1, IP-10, CXCL-1 [KC], MIP-1α, RANTES, IFN-β, IFN-γ, and MX-1), genes encoding IL-6, IL-8 (equivalent to mouse CXCL-1 or KC), IL-12, and IFN-γ were also transiently induced in the lungs of experimentally challenged rhesus macaques (12). Since it has been demonstrated that without cocultivation with plasmacytoid dendritic cells, conventional dendritic cells, or macrophages, human airway epithelial cells exhibit a minimal capacity to induce inflammatory responses and fail to produce IFN-β in response to either SARS-CoV or MERS-CoV infection (32–36), such prompt pulmonary inflammatory responses seen in infected Tg+ mice must be dependent on the interplay among alveolar macrophages, residential dendritic cells, airway epithelial cells, alveolar pneumocytes, and various cells that infiltrated the inflamed lungs. Many genes related to acute inflammatory responses (with IFN-β- and IFN-γ-encoded genes being the exception) were also significantly elevated concurrently with the intense viral replication in the brains of Tg+ mice at 4 dpi (Fig. 3 and 5). However, in contrast to the associated cellular infiltration in the lungs, significant cellular infiltrations were not observed in the infected brains. Although the exact mechanism for the absence of readily detectable cellular infiltrates within the infected brains remains unknown, the acute onset of lethality within 5 to 6 dpi, along with the undetectable viral infection in the brains until 4 dpi, might prevent the inflammatory cascades from occurring. Since mouse neurons, astrocytes, and microglia were permissive to both MERS-CoV and SARS-CoV infection (Fig. 3G) (15) and capable of producing many cytokines and chemokines in response to mouse hepatitis virus infection (37), it is likely that they are the source of the elevated cytokine and chemokine responses in the brain of Tg+ mice. Although the intensities of inflammatory secretions of infected Tg+ mice were positively correlated with the magnitudes of viral infection within the lung and brain, we also noted that higher levels of IP-10 (CXCL10), MCP-1, TNF-α, and RANTES were detected in the brain at 4 dpi than in the lung at 2 dpi, despite the significantly lower virus titers in the lung at 2 dpi, which may relate to the superb ability of brain cells to activate genes encoding inflammatory mediators in response to acute MERS-CoV infection. However, the specific effect of such a dynamic antiviral and inflammatory response on the pathogenesis of MERS-CoV infection in Tg+ mice remains to be fully investigated.
In summary, we provide data in this report of our effort and success in establishing transgenic mice expressing hCD26/DPP4 as a small-animal model for MERS-CoV infection and disease. Not only can these Tg+ mice support a robust MERS-CoV infection, they also develop clinical manifestations and death within a week after infection. We believe that this newly established transgenic mouse model will be useful as an additional model for studying the pathogenesis of and evaluating the efficacy of preventive and therapeutic agents for MERS-CoV infection and disease.
ACKNOWLEDGMENTS
We thank Heinz Feldmann (National Institutes of Health, Hamilton, MT) and Ron A. Fouchier (Erasmus Medical Center, Rotterdam, Netherlands) for providing the MERS-CoV/EMC-2012 strain. We thank Ralph Baric and Amy Sims (University of North Carolina, Chapel Hill, NC) for providing the recombinant RFP-MERS-CoV strain. We also thank Chikao Morimoto and Ohnuma Kei (University of Tokyo, Tokyo, Japan) for providing the pSRa-CD26.2 plasmid used to make the hCD26 transgene construct.
This research was supported in part by National Institutes of Health grant R21AI113206-01 (C.-T.K.T.) and a pilot grant from the Center for Biodefense and Emerging Infectious Diseases, University of Texas Medical Branch, Galveston, TX (C-T.K.T.).
REFERENCES
- 1.Peiris JS, Guan Y, Yuen KY. 2004. Severe acute respiratory syndrome. Nat Med 10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 3.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Assiri A, McGeer A, Perl TM, Price CS, Al Rabeeah AA, Cummings DA, Alabdullatif ZN, Assad M, Almulhim A, Makhdoom H, Madani H, Alhakeem R, Al-Tawfiq JA, Cotten M, Watson SJ, Kellam P, Zumla AI, Memish ZA. 2013. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 369:407–416. doi: 10.1056/NEJMoa1306742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guery B, Poissy J, el Mansouf L, Sejourne C, Ettahar N, Lemaire X, Vuotto F, Goffard A, Behillil S, Enouf V, Caro V, Mailles A, Che D, Manuguerra JC, Mathieu D, Fontanet A, van der Werf S. 2013. Clinical features and viral diagnosis of two cases of infection with Middle East respiratory syndrome coronavirus: a report of nosocomial transmission. Lancet 381:2265–2272. doi: 10.1016/S0140-6736(13)60982-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reusken CB, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, Meyer B, Muth D, Raj VS, Smits-De Vries L, Corman VM, Drexler JF, Smits SL, El Tahir YE, De Sousa R, van Beek J, Nowotny N, van Maanen K, Hidalgo-Hermoso E, Bosch BJ, Rottier P, Osterhaus A, Gortazar-Schmidt C, Drosten C, Koopmans MP. 2013. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis 13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3:e00473–12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coleman CM, Matthews KL, Goicochea L, Frieman MB. 2014. Wild-type and innate immune-deficient mice are not susceptible to the Middle East respiratory syndrome coronavirus. J Gen Virol 95:408–412. doi: 10.1099/vir.0.060640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Wit E, Prescott J, Baseler L, Bushmaker T, Thomas T, Lackemeyer MG, Martellaro C, Milne-Price S, Haddock E, Haagmans BL, Feldmann H, Munster VJ. 2013. The Middle East respiratory syndrome coronavirus (MERS-CoV) does not replicate in Syrian hamsters. PLoS One 8:e69127. doi: 10.1371/journal.pone.0069127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raj VS, Smits SL, Provacia LB, van den Brand JM, Wiersma L, Ouwendijk WJ, Bestebroer TM, Spronken MI, van Amerongen G, Rottier PJ, Fouchier RA, Bosch BJ, Osterhaus AD, Haagmans BL. 2014. Adenosine deaminase acts as a natural antagonist for dipeptidyl peptidase 4-mediated entry of the Middle East respiratory syndrome coronavirus. J Virol 88:1834–1838. doi: 10.1128/JVI.02935-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scobey T, Yount BL, Sims AC, Donaldson EF, Agnihothram SS, Menachery VD, Graham RL, Swanstrom J, Bove PF, Kim JD, Grego S, Randell SH, Baric RS. 2013. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc Natl Acad Sci U S A 110:16157–16162. doi: 10.1073/pnas.1311542110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Wit E, Rasmussen AL, Falzarano D, Bushmaker T, Feldmann F, Brining DL, Fischer ER, Martellaro C, Okumura A, Chang J, Scott D, Benecke AG, Katze MG, Feldmann H, Munster VJ. 2013. Middle East respiratory syndrome coronavirus (MERS-CoV) causes transient lower respiratory tract infection in rhesus macaques. Proc Natl Acad Sci U S A 110:16598–16603. doi: 10.1073/pnas.1310744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falzarano D, de Wit E, Feldmann F, Rasmussen AL, Okumura A, Peng X, Thomas MJ, van Doremalen N, Haddock E, Nagy L, LaCasse R, Liu T, Zhu J, McLellan JS, Scott DP, Katze MG, Feldmann H, Munster VJ. 2014. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLoS Pathog 10:e1004250. doi: 10.1371/journal.ppat.1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J, Li K, Wohlford-Lenane C, Agnihothram SS, Fett C, Gale MJ Jr, Baric RS, Enjuanes L, Gallagher T, McCray PB Jr, Perlman S. 2014. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci U S A 111:4970–4975. doi: 10.1073/pnas.1323279111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tseng CT, Huang C, Newman P, Wang N, Narayanan K, Watts DM, Makino S, Packard MM, Zaki SR, Chan TS, Peters CJ. 2007. Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human Angiotensin-converting enzyme 2 virus receptor. J Virol 81:1162–1173. doi: 10.1128/JVI.01702-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka T, Camerini D, Seed B, Torimoto Y, Dang NH, Kameoka J, Dahlberg HN, Schlossman SF, Morimoto C. 1992. Cloning and functional expression of the T cell activation antigen CD26. J Immunol 149:481–486. [PubMed] [Google Scholar]
- 17.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 18.Wakamiya M, Lindsay EA, Rivera-Perez JA, Baldini A, Behringer RR. 1998. Functional analysis of Gscl in the pathogenesis of the DiGeorge and velocardiofacial syndromes. Hum Mol Genet 7:1835–1840. doi: 10.1093/hmg/7.12.1835. [DOI] [PubMed] [Google Scholar]
- 19.Tao X, Mei F, Agrawal A, Peters CJ, Ksiazek TG, Cheng X, Tseng CT. 2014. Blocking of exchange proteins directly activated by cAMP leads to reduced replication of Middle East respiratory syndrome coronavirus. J Virol 88:3902–3910. doi: 10.1128/JVI.03001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tseng CT, Tseng J, Perrone L, Worthy M, Popov V, Peters CJ. 2005. Apical entry and release of severe acute respiratory syndrome-associated coronavirus in polarized Calu-3 lung epithelial cells. J Virol 79:9470–9479. doi: 10.1128/JVI.79.15.9470-9479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J, Chu H, Li C, Wong BH, Cheng ZS, Poon VK, Sun T, Lau CC, Wong KK, Chan JY, Chan JF, To KK, Chan KH, Zheng BJ, Yuen KY. 2014. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis 209:1331–1342. doi: 10.1093/infdis/jit504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshikawa N, Yoshikawa T, Hill T, Huang C, Watts DM, Makino S, Milligan G, Chan T, Peters CJ, Tseng CT. 2009. Differential virological and immunological outcome of severe acute respiratory syndrome coronavirus infection in susceptible and resistant transgenic mice expressing human angiotensin-converting enzyme 2. J Virol 83:5451–5465. doi: 10.1128/JVI.02272-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Doremalen N, Miazgowicz KL, Milne-Price S, Bushmaker T, Robertson S, Scott D, Kinne J, McLellan JS, Zhu J, Munster VJ. 2014. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 88:9220–9232. doi: 10.1128/JVI.00676-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts A, Lamirande EW, Vogel L, Jackson JP, Paddock CD, Guarner J, Zaki SR, Sheahan T, Baric R, Subbarao K. 2008. Animal models and vaccines for SARS-CoV infection. Virus Res 133:20–32. doi: 10.1016/j.virusres.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, Herman BD, Sheahan T, Heise M, Genrich GL, Zaki SR, Baric R, Subbarao K. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog 3:e5. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambeir AM, Durinx C, Scharpe S, De Meester I. 2003. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci 40:209–294. doi: 10.1080/713609354. [DOI] [PubMed] [Google Scholar]
- 27.Yu DM, Yao TW, Chowdhury S, Nadvi NA, Osborne B, Church WB, McCaughan GW, Gorrell MD. 2010. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J 277:1126–1144. doi: 10.1111/j.1742-4658.2009.07526.x. [DOI] [PubMed] [Google Scholar]
- 28.Mu J, Petrov A, Eiermann GJ, Woods J, Zhou YP, Li Z, Zycband E, Feng Y, Zhu L, Roy RS, Howard AD, Li C, Thornberry NA, Zhang BB. 2009. Inhibition of DPP-4 with sitagliptin improves glycemic control and restores islet cell mass and function in a rodent model of type 2 diabetes. Eur J Pharmacol 623:148–154. doi: 10.1016/j.ejphar.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 29.Takasawa W, Ohnuma K, Hatano R, Endo Y, Dang NH, Morimoto C. 2010. Inhibition of dipeptidyl peptidase 4 regulates microvascular endothelial growth induced by inflammatory cytokines. Biochem Biophys Res Commun 401:7–12. doi: 10.1016/j.bbrc.2010.08.112. [DOI] [PubMed] [Google Scholar]
- 30.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 31.Gralinski LE, Baric RS. 2014. Molecular pathology of emerging coronavirus infections. J Pathol 235:185–195. doi: 10.1002/path.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, Thiel V, Ludewig B. 2007. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 109:1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan RW, Chan MC, Agnihothram S, Chan LL, Kuok DI, Fong JH, Guan Y, Poon LL, Baric RS, Nicholls JM, Peiris JS. 2013. Tropism of and innate immune responses to the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J Virol 87:6604–6614. doi: 10.1128/JVI.00009-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kindler E, Jonsdottir HR, Muth D, Hamming OJ, Hartmann R, Rodriguez R, Geffers R, Fouchier RA, Drosten C, Muller MA, Dijkman R, Thiel V. 2013. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio 4:e00611–12. doi: 10.1128/mBio.00611-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiss SR, Navas-Martin S. 2005. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zielecki F, Weber M, Eickmann M, Spiegelberg L, Zaki AM, Matrosovich M, Becker S, Weber F. 2013. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J Virol 87:5300–5304. doi: 10.1128/JVI.03496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rempel JD, Quina LA, Blakely-Gonzales PK, Buchmeier MJ, Gruol DL. 2005. Viral induction of central nervous system innate immune responses. J Virol 79:4369–4381. doi: 10.1128/JVI.79.7.4369-4381.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brinster RL, Allen JM, Behringer RR, Gelinas RE, Palmiter RD. 1988. Introns increase transcriptional efficiency in transgenic mice. Proc Natl Acad Sci U S A 85:836–840. doi: 10.1073/pnas.85.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmiter RD, Sandgren EP, Avarbock MR, Allen DD, Brinster RL. 1991. Heterologous introns can enhance expression of transgenes in mice. Proc Natl Acad Sci U S A 88:478–482. doi: 10.1073/pnas.88.2.478. [DOI] [PMC free article] [PubMed] [Google Scholar]